一种盐酸溴己新口服溶液中降解杂质的检测方法与流程
1.本发明涉及药物检测技术领域,特别是涉及一种盐酸溴己新口服溶液中降解杂质的检测方法。
背景技术:
2.盐酸溴己新口服溶液在临床上用于急性及慢性支气管炎、哮喘、支气管扩张、肺气肿,尤适用于白色粘痰咳出困难者及因痰液广泛阻塞小支气管引起的危重急症等。盐欧洲药典7.0版收载了5种降解杂质(a、b、c、d、e),其中杂质e为盐酸溴己新口服溶液剂中的主要杂质,盐酸溴己新口服溶液中降解杂质的含量对药效影响很大,因此,准确检测盐酸溴己新口服溶液中杂质的含量,对保证盐酸溴己新口服溶液产品质量,保证用药安全具有重要的研究意义。
3.目前,各国药典中均未见收载关于盐酸溴己新口服溶液的有关物质检测方法。酸溴己新口服溶液的有效成分盐酸溴己新在贮存时会发生缓慢的降解,为了提高稳定性,盐酸溴己新口服溶液中通常加入有防腐剂、稳定剂和增粘剂等辅料,这些辅料对降解杂质的检测具有一定的干扰性。
4.现有的盐酸溴己新原料药、盐酸溴己新片和盐酸溴己新注射液的有关物质方法,均不能有效分离盐酸溴己新口服溶液中防腐剂苯甲酸钠和降解杂质e。盐酸溴己新口服溶液中含有比例高达20~30%的麦芽糖醇和羟乙基纤维素,麦芽糖醇和羟乙基纤维素极易溶于水,不溶于高比例有机溶剂。在采用色谱法检测盐酸溴己新口服溶液中降解杂质时,如流动相采用了高有机相,此类成分会在色谱柱中析出,快速的损坏色谱柱,无法顺利完成检测。
5.例如,《中国药典》2020年版二部收载了盐酸溴己新原料药和盐酸溴己新片剂的有关物质方法,流动相中有机相比例为80%,盐酸溴己新口服溶液中的大量麦芽糖醇会析出,容易堵塞和损害色谱柱。
6.例如,申请公布号:cn 107703230 a,中国专利公开了一种盐酸溴己新有关物质的高效液相色谱,流动相的ph为5.2,不利于盐酸溴己新口服溶液中苯甲酸钠与降解杂质e的分离,且梯度运行时间太长(80min)。
7.例如,申请公布号:cn 103499647 a,中国专利公开了一种盐酸溴己新葡萄糖注射液的中间体或成品的检测方法,其流动相的ph为3.3,不利于盐酸溴己新口服溶液中苯甲酸钠与降解杂质e的分离,且能分离的杂质只有4个。
8.研究发现,供试品处理方式对色谱柱的寿命有很大的影响,且供试品溶液的稳定性均低于24h,为确保检测结果的准确性,需要采用即配即测,且一般需要采用低温进样,无法实现批量化快速检测。
技术实现要素:
9.鉴于以上所述现有技术的缺点,本发明的目的在于提供一种盐酸溴己新口服溶液
中降解杂质的检测方法,用于解决现有盐酸溴己新口服溶液中降解杂质的检测过程中存在着干扰性强、分离效果差、部分辅料损坏色谱柱、检测时间长、检测结果不准确的问题。
10.为实现上述目的及其他相关目的,本发明提供一种盐酸溴己新口服溶液中降解杂质的检测方法,采用高效液相色谱法检测,供试品溶液采用乙腈溶解处理;以十八烷基硅烷键合硅胶为填充剂,以ph为6.5~7.5的缓冲盐溶液浓度为流动相a,以甲醇乙腈混合液为流动相b,进行梯度洗脱。
11.本发明通过优化组合不同的色谱条件,采用ph为6.5~7.5的缓冲盐溶液浓度作为流动相a及低温进样,可使供试品溶液在48小时内降解杂质的量基本不变,故不需临用新制,这使得更为高效的批量检测成为可能;采用甲醇乙腈混合液作为流动相b可以消除检测过程中辅料溶液产生的严重干扰,有效提高了本品各杂质与主成分间的分离效果,实现了复杂体系中微量杂质的准确快速定量检测。按照现有技术中的供试品处理方法,色谱柱进样不到10针后出现堵塞和塌陷的现象,本发明中供试品溶液制备时先用乙腈溶解并过滤再定容,有效去除了辅料中大量的不溶性辅料,从而大大提高了色谱柱的使用寿命。
12.优选地,梯度洗脱程序为:
[0013]0→
8min,流动相a体积百分含量:流动相b体积百分含量为45%:55%
→
55%:45%;
[0014]8→
30min,流动相a体积百分含量:流动相b体积百分含量为45%:55%
→
55%:45%;
[0015]
30
→
55min,流动相a体积百分含量:流动相b体积百分含量为5%:85%
→
15%:95%;
[0016]
55
→
56min,流动相a体积百分含量:流动相b体积百分含量为5%:85%
→
15%:95%;
[0017]
56
→
65min,流动相a体积百分含量:流动相b体积百分含量为45%:55%
→
55%:45%。
[0018]
上述洗脱程序也可以如表1所示:
[0019]
表1.梯度洗脱程序
[0020]
时间(分钟)流动相a(%)流动相b(%)0
→
845~5555~458
→
3045~5555~4530
→
555~1585~9555
→
565~1585~9556
→
6545~5555~45
[0021]
本发明通过优化梯度洗脱程序并结合色谱条件,可以使得主峰峰型对称性好,各杂质之间,杂质与主峰之间分离度好,空白辅料不干扰杂质的检测。
[0022]
更优选地,梯度洗脱程序为:
[0023]0→
8min,流动相a体积百分含量:流动相b体积百分含量为50%:50%;
[0024]8→
30min,流动相a体积百分含量:流动相b体积百分含量为50%:50%;
[0025]
30
→
55min,流动相a体积百分含量:流动相b体积百分含量为10%:90%;
[0026]
55
→
56min,流动相a体积百分含量:流动相b体积百分含量为10%:90%;
[0027]
56
→
65min,流动相a体积百分含量:流动相b体积百分含量为50%:50%。
[0028]
上述洗脱程序也可以如表2所示:
[0029]
表2.梯度洗脱程序
[0030]
时间(分钟)流动相a(%)流动相b(%)0
→
850508
→
30505030
→
55109055
→
56109056
→
655050。
[0031]
优选地,所述缓冲盐溶液为浓度为0.01mol/l的乙酸铵溶液。采用乙酸或氨水调整乙酸铵溶液的ph值。
[0032]
优选地,所述甲醇乙腈混合液中甲醇与乙腈的体积比为1:4。采用该配比的甲醇乙腈混合液作为流动相b可以消除检测过程中辅料溶液产生的严重干扰,有效提高了本品各杂质与主成分间的分离效果,实现了复杂体系中微量杂质的准确快速定量检测。
[0033]
优选地,所述高效液相色谱使用245nm的紫外检测波长,柱温为35~45℃,流速为0.8~1.2ml/min。
[0034]
优选地,柱温为40℃,流速为1.0ml/min。
[0035]
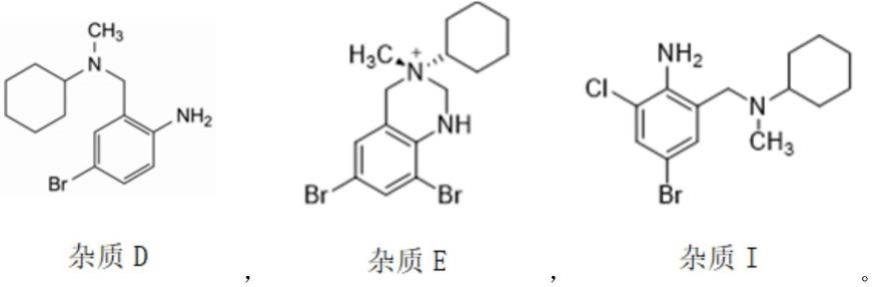
优选地,所述降解杂质选自以下杂质中的一种或几种:
[0036][0037][0038]
优选地,所述杂质a的校正因子为0.69;所述杂质b的校正因子为0.48,所述杂质c的校正因子为0.91,所述杂质d的校正因子为0.81,所述杂质e的校正因子为0.97,所述杂质i的校正因子为0.99。
[0039]
优选地,所述盐酸溴己新口服溶液中降解杂质的检测方法,包括以下步骤:
[0040]
1)取5ml盐酸溴己新口服溶液,加入3ml乙腈溶解,滤过,用乙腈将滤液稀释至
10ml,作为供试品溶液;
[0041]
2)取空白辅料溶液5ml,加3ml乙腈溶解,滤过,用乙腈将滤液稀释至10ml,作为空白辅料测定液;
[0042]
3)对照品溶液配制:
[0043]
分别称取各降解杂质,加乙腈溶解,制成浓度为0.2mg/ml的降解杂质乙腈溶液,作为降解杂质对照品贮备溶液;
[0044]
取盐酸溴己新对照品,加乙腈溶解,制成浓度为0.2mg/ml的盐酸溴己新乙腈溶液,作为盐酸溴己新对照品贮备溶液;
[0045]
称取各降解杂质对照品贮备溶液和盐酸溴己新对照品贮备溶液混合,用乙腈稀释为各组分浓度均为0.02mg/ml的混合液,作为混合杂质对照品贮备溶液;
[0046]
将混合杂质对照品贮备溶液稀释25倍,作为混合对照品溶液;
[0047]
5)定位溶液配制:
[0048]
分别量取1ml各降解杂质对照品贮备溶液,用乙腈稀释为浓度为0.008mg/ml的降解杂质乙腈溶液,作为降解杂质定位溶液;
[0049]
取1ml盐酸溴己新对照品贮备溶液,用乙腈稀释为浓度为0.008mg/ml的盐酸溴己新乙腈溶液,作为盐酸溴己新定位溶液;
[0050]
6)加标样品溶液配制:精密量取5ml供试品,加3ml乙腈溶解,滤过,滤液置10ml量瓶中,加0.8ml混合杂质对照品贮备溶液,用乙腈稀释至刻度,摇匀,取续滤液,即得加标样品溶液;
[0051]
7)取供试品溶液、空白辅料测定液,对照品溶液、定位溶液和加标样品溶液,分别注入液相色谱仪,按照所述色谱条件进行测定,记录色谱图,根据供试品溶液的色谱图和混合对照品溶液的色谱图,确定供试品溶液中降解杂质的限度。
[0052]
如上所述,本发明的盐酸溴己新口服溶液中降解杂质的检测方法,具有以下有益效果:通过优化组合不同的色谱条件,采用ph为6.5~7.5的缓冲盐溶液浓度作为流动相a及5~8℃的低温进样,可使供试品溶液在48小时内降解杂质的量基本不变,故不需临用新制,这使得更为高效的批量检测成为可能;采用甲醇乙腈混合液作为流动相b可以消除检测过程中辅料溶液产生的严重干扰,有效提高了本品各杂质与主成分间的分离效果,实现了复杂体系中微量杂质的准确快速定量检测。按照现有技术中的供试品处理方法,色谱柱进样不到10针后出现堵塞和塌陷的现象,本发明中供试品溶液制备时先用乙腈溶解并过滤再定容,有效去除了辅料中大量的不溶性辅料,从而大大提高了色谱柱的使用寿命。
附图说明
[0053]
图1显示为本发明盐酸溴己新口服溶液中检测到的降解杂质及空白辅料叠加hplc图。
具体实施方式
[0054]
以下通过特定的具体实例说明本发明的实施方式,本领域技术人员可由本说明书所揭露的内容轻易地了解本发明的其他优点与功效。本发明还可以通过另外不同的具体实施方式加以实施或应用,本说明书中的各项细节也可以基于不同观点与应用,在没有背离
本发明的精神下进行各种修饰或改变。
[0055]
须知,本发明的实施例中所使用的试剂均为色谱纯,除非另有说明。
[0056]
1.试剂及材料:
[0057]
乙酸铵、氨水、磷酸、乙腈、甲醇。
[0058]
盐酸溴己新对照品(来源:中国食品药品检定研究院,批号:100427-201903,含量:99.8%);
[0059]
杂质a对照品(来源:hst,批号:hst-b3801-202005,含量:97.16%);
[0060]
杂质b对照品(来源:hst,批号:hst-b3802-202005,含量:98.73%);
[0061]
杂质c对照品(来源:hst,批号:hst-b3803-202006,含量:99.05%);
[0062]
杂质d对照品(来源:hst,批号:hst-b3804-202006,含量:97.36%);
[0063]
杂质e对照品(来源:hst,批号:hst-b3805-202006,含量:97.34%);
[0064]
盐酸溴己新对照品(来源:中国食品药品检定研究院,批号:101387-201501,含量:100%);空白辅料(来源:自研,批号:k0721080303)
[0065]
盐酸溴己新口服溶液(来源:浙江康恩贝制药股份有限公司,批号:y210401)。
[0066]
2.主要仪器:
[0067]
waters e2695高效液相色谱仪,赛多利斯sqp电子天平,梅特勒fe-28ph计。
[0068]
3.色谱条件:
[0069]
色谱柱:用十八烷基硅烷键合硅胶为填充剂(ultimate xb-c18 4.6
×
250mm,5μm)
[0070]
流速:1.0ml/min;柱温:40℃;波长:245nm;进样温度5℃,进样量:20μl。
[0071]
4.流动相及梯度洗脱程序:
[0072]
以ph为6.8,浓度为0.01mol/l乙酸铵溶液作为流动相a,以甲醇乙腈按照体积比1:4的混合液作为流动相b,按照表2所示的梯度洗脱程序进行洗脱:
[0073]
表2.梯度洗脱程序
[0074]
时间(分钟)流动相a(%)流动相b(%)0
→
850508
→
30505030
→
55109055
→
56109056
→
655050。
[0075]
5.方法学验证:
[0076]
5.1专属性
[0077]
通过对照品定位溶液进行杂质确认和归属,采用空白辅料溶液排除辅料对杂质检测干扰。
[0078]
(1)溶液配制
[0079]
各溶液的配制方法如表3所示:
[0080]
表3.各溶液的配制方法
[0081]
[0082]
[0083][0084]
专属性试验结果如图1和表4所示,图1为本发明盐酸溴己新口服溶液中检测到的降解杂质及空白辅料叠加hplc图。
[0085]
表4.专属性试验结果
[0086]
[0087][0088]
结合图1和表4可以看出,空白辅料峰在主峰及已知杂质峰出峰处无干扰;各杂质之间及杂质与主峰分离的均大于2.5,表明本发明的检测方法的分离度良好。
[0089]
5.2线性与范围:
[0090]
线性关系以测得的响应信号(峰面积)对被分析物浓度的函数作图,用最小二乘法进行线性回归,要求该线性回归系数r2的数值应不小于0.990。对未知杂质,用盐酸氨溴索对照品替代考察单个未知杂质的线性和范围,盐酸溴己新线性试验结果如表5-表11所示:
[0091]
表5.盐酸溴己新线性试验结果
[0092][0093]
从表5可以看出,盐酸溴己新在0.0833μg/ml~1.6655μg/ml浓度范围内,线性方程为y=28520x+107.58,相关系数r=0.9999,y轴截距为100%响应值的0.47%;盐酸溴己新响应值的rsd为1.8%,线性关系良好。
[0094]
表6.杂质a线性试验结果
[0095]
[0096][0097]
从表6可以看出,杂质a在0.0850μg/ml~1.7007μg/ml浓度范围内,线性方程为y=41462x+102.86,相关系数r=0.9999,y轴截距为100%响应值的0.31%;杂质a响应值的rsd为1.4%,线性关系良好。
[0098]
表7.杂质b线性试验结果
[0099][0100]
从表7可以看出,杂质b在0.0405μg/ml~1.6208μg/ml浓度范围内,线性方程为y=59800x-33.529,相关系数r=0.9999,y轴截距为100%响应值的0.07%;杂质b响应值的rsd为2.5%,线性关系良好。
[0101]
表8.杂质c线性试验结果
[0102][0103][0104]
从表8可以看出,杂质c在0.1296μg/ml~1.3821μg/ml浓度范围内,线性方程为y=
31039x-353.45,相关系数r=0.9997,y轴截距为100%响应值的0.86%;杂质c响应值的rsd为4.4%,线性关系良好。
[0105]
表9.杂质d线性试验结果
[0106][0107]
从表9可以看出,杂质d在0.1046μg/ml~1.3944μg/ml浓度范围内,线性方程为y=35114x+168.9,相关系数r=0.9998,y轴截距为100%响应值的0.60%;杂质d响应值的rsd为4.0%,线性关系良好。
[0108]
表10.杂质e线性试验结果
[0109][0110][0111]
从表10可以看出,杂质e在0.0934μg/ml~3.7362μg/ml浓度范围内,线性方程为y=29542x+80.064,相关系数r=0.9999,y轴截距为100%响应值的0.14%;杂质e响应值的rsd为3.4%,线性关系良好。
[0112]
表11.杂质i线性试验结果
[0113][0114]
从表11可以看出,杂质i在0.0842μg/ml~3.7432μg/ml浓度范围内,线性方程为y=28317x-882.65,相关系数r=0.9996,y轴截距为100%响应值的0.36%;杂质i响应值的rsd为2.2%,线性关系良好。
[0115]
已知降解杂质校正因子
[0116]
本品已知杂质采用标准曲线法测定校正因子,即精密称取杂质对照品和主成分对照品,分别制备系列溶液(涵盖报告限度、标准限度)分别进样,按最小二乘法以供试品浓度对响应因子(峰面积)进行线性回归,求得两条标准曲线,主成分与已知杂质斜率之比即为校正因子,具体参见表12:
[0117]
表12.校正因子测定结果
[0118]
名称斜率校正因子盐酸溴己新28520/杂质a414620.69杂质b598000.48杂质c310390.91杂质d351140.81杂质e295420.97杂质i283170.99
[0119]
5.4检测限及定量限
[0120]
对于已知杂质,检测限(lod)和定量限(loq)是根据信噪比法来确定的。把已知浓度的杂质储备溶液稀释到低浓度的试样,测出的信号与空白处的信号(基线噪音)进行比较,算出能被可靠的检测出的最低浓度或百分比,检测限与定量限均应低于报告限度(0.05%)。
[0121]
对未知杂质,用盐酸氨溴索对照品替代考察单个未知杂质的定量限和检测限,结果分别如表13和表14所示。
[0122]
对各杂质与盐酸氨溴索对照品的混合溶液,逐级稀释,根据信噪比s/n≥10确定loq,根据s/n≥3确定lod。
[0123]
表13.定量限结果
[0124][0125]
表14.检测限结果
[0126][0127]
5.5准确度试验
[0128]
准确度是通过在供试品中加入杂质限度的50%、100%及150%三个不同浓度各已知杂质测定的回收率所得。已知杂质的准确度是加入已知量的杂质,再测定加样样品中已知杂质的试验结果和理论值之间的比值(回收率),以百分比%表达,要求每份样品的回收率在5.0%~105.0%之间,rsd≤5%。准确度试验结果如表15所示:
[0129]
表15.准确度试验结果
[0130]
[0131][0132]
从表15可以看出,杂质a的回收率均在96.3%~97.8%之间,9份结果的rsd为0.5%。杂质b的回收率均在96.4%~1038%之间,9份结果的rsd为2.7%。杂质c的回收率均在98.1%~102.5%之间,9份结果的rsd为1.5%。杂质d的回收率均在96.1%~100.0%之间,9份结果的rsd为1.3%。杂质e的回收率均在96.9%~101.8%之间,9份结果的rsd为1.5%。杂质i的回收率均在98.9%~100.9%之间,9份结果的rsd为0.6%。表明本方法回收率良好。
[0133]
5.5重复性试验
[0134]
重复性试验是通过配制6个样品溶液并进行测试,要求6次数据结果杂质变化量的绝对偏差不得超过0.1%。
[0135]
供试品溶液:精密量取盐酸溴己新口服溶液5ml,加乙腈约3ml,滤过,滤液置10ml量瓶中,加乙腈稀释至刻度,摇匀,即得。同法配制6份。
[0136]
计算公式:
[0137][0138]
其中:a供:供试品峰面积;d供:供试品稀释倍数;
[0139]
l:规格(含盐酸溴己新4mg/5ml或8mg/5ml);1000:单位换算;
[0140]
f:响应因子;a对:对照品峰面积;d对:对照品稀释倍数;
[0141]
m对:对照品称样量,mg;h对:对照品纯度。
[0142]
备注:已知杂质按杂质对照品溶液响应值计算,未知杂质按盐酸溴己新响应值计算。
[0143]
重复性检测结果如表16所示:
[0144]
表16.重复性检测结果
[0145]
乙腈(60:40)。
[0161]
检测结果显示:第一个杂质与空白溶剂峰之间不能达到基线分离,且部分已知杂质不能达到基线分离。
[0162]
对比例4
[0163]
对比例4与本技术实施例的区别在于,流动相及洗脱梯度不同:参照专利公开号cn107703230a,一种盐酸溴己新有关物质的高效液相色谱,ph为5~5.5的缓冲盐溶液为流动相a,甲醇为流动相b,表18所示的洗脱梯度:
[0164]
表18.对比例4的洗脱梯度
[0165]
时间(分钟)流动相a(%)流动相b(%)0
→
30653530
→
60406060
→
70158570
→
71158571
→
806535。
[0166]
检测结果显示:主峰峰型对称性差,运行时间太长。
[0167]
综上所述,采用本技术特定色谱条件的检测方法,得到的谱图主峰峰型对称性好,各杂质之间,杂质与主峰之间分离度好,空白辅料不干扰杂质,优于对比例1-4的检测结果。
[0168]
上述实施例仅例示性说明本发明的原理及其功效,而非用于限制本发明。任何熟悉此技术的人士皆可在不违背本发明的精神及范畴下,对上述实施例进行修饰或改变。因此,举凡所属技术领域中具有通常知识者在未脱离本发明所揭示的精神与技术思想下所完成的一切等效修饰或改变,仍应由本发明的权利要求所涵盖。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1