一种墨迹中溶剂成分的检测方法
1.本发明涉及文件材料检验技术领域,尤其是一种墨迹中溶剂成分的检测方法。
背景技术:
2.书写墨迹中墨水的分析鉴定一直是文件材料检验学的热点问题,包括墨水种类的鉴定和书写时间的鉴定。合同、借据等书证签名的真伪,内容是否有添改,书写的时间等问题都涉及对墨迹成分的分析鉴定。研究墨迹中挥发性溶剂的种类及其残留量随时间变化的规律,是墨迹书写时间鉴定的重要手段。
3.圆珠笔墨水中常见的溶剂成分主要有苯甲醇、2-苯氧乙醇、二甘醇乙醚,中性、水性笔墨水中常见的溶剂成分主要有乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇等。
4.近年来文献报道的多元醇类定性定量分析方法主要采用了气相色谱法和气相色谱-质谱联用法。因为多元醇类作为保湿剂使用的广泛性,烟草化学行业已经建立了部分多元醇的定性定量分析方法(如文献[1]段沅杏,吴亿勤,杨威,等.gc/ms法同时测定电子烟烟气释放物中的5种醇类化合物[j].烟草科技.2015,48(10):43-47;文献[2]张杰,李鹏,孙世豪,等.gc/ms法同时检测无烟气烟草制品中的1,2-丙二醇、丙三醇和三甘醇[j].烟草科技.2011(03):36-42;中国专利申请cn111272938a)。虽然墨水中也存在部分醇类溶剂,但由于应用范围不同,墨水中的常见溶剂成分如乙二醇、1,3-丙二醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚等,在烟草化学行业的多元醇检测方法中未见研究数据,这些成分用现有的方法不一定能完全分离。此外,由于样品种类差异巨大,样品的前处理和内标物的选择也有很大差别,因此烟草化学行业的多元醇类检测方法不能直接应用于墨迹中溶剂成分的分析。
[0005]
在文件检验研究领域,定性测定多元醇种类的文献较多,定量测定其含量的文献较少,而且定量测定多以样本自身的相对含量来表征,其结果不便于同行研究者进行横向比较印证。如郭东东等(郭东东,吕荫妮,张海鹏.黑色水性笔、中性笔墨迹中溶剂成分的gc分析[j].福建分析测试.2015,24(04):48-52.),牛凡等(牛凡,黄建同,何森,等.用气相色谱-质谱法和薄层色谱法分析蓝色圆珠笔油墨及其分类[j].理化检验(化学分册).2017,53(01):22-27.),司法鉴定技术规范sf/z jd0203004-2018报道了采用气相色谱法、气相色谱-质谱法等方法对签字笔油墨中溶剂的组成进行了种类分析,未进行定量研究。li等(li b,xie p,guo y,et al.gc analysis of black gel pen ink stored under different conditions[j].journal of forensic sciences.2014,59(2):543-549.),ni等(ni y,he n,l
ü
y,et al.study of ink aging:targeting triethylene glycol in carbon-based black gel ink strokes on paper[j].forensic science international.2020,311:110296.)报道了通过签字笔字迹中墨水溶剂残留量,判断书写时间,分别研究了字迹中乙二醇、二甘醇、丙三醇、1,2-丙二醇、三甘醇的残留量与书写时间的关系,但仅是测定了各溶剂成分相对于内标的比值,其他研究者无法得知其绝对含量的大小。然而,不同笔的溶剂成分的初始含量差别很大,相对含量所体现的有效信息较少,测定其绝对含量对于解释墨迹
书写时间的鉴定结果具有重要参考价值。
[0006]
仅有少数文献对墨迹中溶剂的含量进行了绝对定量分析,但都有一定缺陷:(1)对不同种类的笔迹测定有局限性,如koenig等(koenig a,magnolon s,weyermann c.a comparative study of ballpoint ink ageing parameters using gc/ms[j].forensic science international.2015,252:93-106.),李双萍等(李双萍,杨旭,孙其然.gc/ms法对圆珠笔墨迹中苯甲醇和苯氧乙醇的定量分析[j].中国司法鉴定.2020(03):41-45.)对圆珠笔油墨的溶剂成分进行了定量测定,但其方法只能适用于圆珠笔油墨中溶剂成分的测定,不能用于其它种类的笔;(2)分析的溶剂成分的种类不够全面,如郭东东等(郭东东,吕荫妮,张海鹏.溶剂提取-工作曲线法分析黑色墨迹的形成时间[j].中国刑警学院学报.2015(02):76-78.)对不同形成时间的中性笔、水性笔墨迹中的乙二醇、丙三醇的溶剂量进行了分析,而没有对二甘醇、三甘醇、2-苯氧乙醇等其它常见溶剂成分进行分析;(3)分析检测耗时较长,通常在30min左右。
[0007]
因此,需要提供一种墨迹中溶剂成分的检测方法,兼具高准确度、高精密度,且耗时短,可同时检测多种溶剂成分。
技术实现要素:
[0008]
本发明的目的在于,克服现有技术中的缺陷,提供一种墨迹中溶剂成分的检测方法,使用配有fid检测器的气相色谱仪对多种墨迹进行检测,兼具高灵敏度、高准确度、高精密度,且耗时短,可同时检测多种溶剂成分。
[0009]
为实现上述目的,本发明采用如下技术方案:
[0010]
一种墨迹中溶剂成分的检测方法,包括如下步骤:
[0011]
s1.配制混合标准溶液:
[0012]
用内标物和目标化合物的标准品配制不同浓度的混合标准溶液;
[0013]
所述内标物为1,3-丁二醇或1,4-丁二醇;所述目标化合物为乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇和二甘醇乙醚;
[0014]
s2.制备样品溶液:
[0015]
使用取样器对待测样品取样,取得的样品浸没于含内标物的提取剂中,提取后进行离心,取上清液即为样品溶液;
[0016]
s3.气相色谱分析:
[0017]
对所述混合标准溶液和样品溶液进行气相色谱分析,采用气相色谱法对墨迹中溶剂成分进行定性检测或定量检测。
[0018]
本发明的检测方法中,为了消除进样量和仪器不稳定因素造成的误差,采用内标法配合气相色谱分析,对书写有墨迹的样品进行定性或定量分析检测。
[0019]
本发明以1,3-丁二醇或1,4-丁二醇为内标物,采用内标法进行定量分析。1,3-丁二醇与1,4-丁二醇均不常用于墨水溶剂,作为内标物可以与待检测的目标化合物完全分离。此外,本发明采用的内标物与目标化合物具有接近的物理化学性质,且在本发明的气相色谱法中,内标物的色谱峰位置与目标化合物的色谱峰的位置相近,从而实现了墨迹中溶剂成分的良好定性定量检测。
[0020]
测定多元醇类的现有技术中通常选用的内标物有1,3-丁二醇、1,4-丁二醇、苯甲
酸乙酯等。发明人研究发现,苯甲酸乙酯与本发明中的乙二醇、丙三醇、三甘醇等多数目标物的化学结构差异较大,相同质量浓度的苯甲酸乙酯在fid检测器上的响应比其它目标物高几倍。此外苯甲酸乙酯挥发性极强,容易造成其它试剂的交叉污染。因此,苯甲酸乙酯在本发明的检测方法中难以作为内标物使用。
[0021]
优选地,所述内标物为1,3-丁二醇。
[0022]
在本发明的气相色谱分析条件下,1,4-丁二醇的出峰时间在二甘醇乙醚与二甘醇之间,混合标准品的色谱图在中段稍显拥挤;而1,3-丁二醇出峰时间合适,十种化合物在色谱图上分布均匀,各化合物的分离度相对最大。因此,以1,3-丁二醇为内标物,可以实现更优的检测结果。
[0023]
通过本发明的检测方法,合理选择内标物、制备样品溶液,并设定适宜的气相色谱分析条件,单个样品的检测耗时仅为12min左右,其中信号采集时间仅为8.8min左右,极大地缩短了检测耗时,大大提高了检测效率。此外,与常规的气相色谱-质谱联用法相比,本发明的方法仅采用气相色谱仪进行检测分析,而无需联用质谱仪检测,在降低运行成本、减少仪器使用的情况下,实现了高准确度、高精密度的检测效果。
[0024]
优选地,步骤s1中,包括如下步骤:
[0025]
将内标物与提取剂混合,得到内标工作液;称取目标化合物的标准品,加至内标工作液中,经稀释,得到不同浓度的混合标准溶液。
[0026]
更优选地,步骤s1中,包括如下步骤:
[0027]
称取内标物的标准品,用提取剂配制浓度为10mg/ml的内标储备液,再用提取剂稀释定容,得到内标物浓度为50μg/ml的内标工作液;
[0028]
分别称取各目标化合物的标准品,用内标工作液配制成10mg/ml的混合标准溶液,然后用内标工作液稀释,配制成1、5、10、50、100、200、300μg/ml共7个级别含50μg/ml内标物的9种目标化合物的混合标准溶液。
[0029]
可选地,所述提取剂为甲醇、乙腈、乙醇、氯仿、n,n-二甲基甲酰胺中的至少一种。
[0030]
优选地,所述提取剂为甲醇。
[0031]
在本发明中,所述使用取样器对待测样品取样是指:使用打孔器对书写有墨迹的待测样品进行打孔取样。
[0032]
优选地,所述打孔器的打孔直径为0.1~1mm。
[0033]
更优选地,所述打孔器的打孔直径为0.5mm。
[0034]
通过打孔器对待测样品进行打孔取样,得到一定直径的圆片为取得的样品,再进行后续提取。
[0035]
可选地,所述提取的方式可以为静置、机械振荡、超声中的至少一种。
[0036]
通过提取,待测样品的墨迹中含有的溶剂逐渐溶解至含内标物的提取剂中,再经过离心,取上清液,从而获得提取有目标化合物的样品溶液。
[0037]
优选地,步骤s2中,所述提取为在10~50℃下静置5~60min。
[0038]
更优选地,步骤s2中,所述提取为在25℃下静置40min。
[0039]
以甲醇为提取剂,通过对提取的温度和时间进行研究,发明人发现,在25℃,40min的条件下静置提取样品,对大多数溶剂成分的提取率最高。
[0040]
优选地,步骤s3中,所述气相色谱分析条件为:
[0041]
色谱柱:db-alc2色谱柱,规格是30m
×
0.32mm
×
1.2μm;
[0042]
载气:氮气,纯度99.999%;
[0043]
柱流量:恒流模式1~3ml/min;
[0044]
进样口温度230~290℃,不分流进样,吹扫时间0.3~1.0min,吹扫流量30~70ml/min;
[0045]
柱温升温程序:
[0046]
初始温度40~85℃保持0.5~5min,以15~130℃/min升温至110~135℃保持2~4min,以15~130℃/min升温至150~190℃保持1~4min,以15~130℃/min升温至220~260℃保持2~15min,后运行:在220~260℃保持2~15min;
[0047]
或者:初始温度40~85℃保持0.5~5min,然后以15~130℃/min的速率升温到110~135℃并保持2~4min,然后以15~130℃/min的速率升温到220~260℃保持4~15min;
[0048]
检测器温度:250~300℃。
[0049]
本发明选的色谱柱为db-alc2色谱柱,规格是30m
×
0.32mm
×
1.2μm。
[0050]
色谱柱的选择应实现目标物的分离度高、柱效高的效果。发明人通过大量创造性试验研究发现,采用agilent j&w生产的db-alc2色谱柱,在本发明的气相色谱分析条件下,能将9种目标化合物及内标完全分离,且柱效高、峰形尖锐对称,包括后运行的整个气相循环时间仅需12min。
[0051]
而其他常规的用于醇类分析的色谱柱均有不同程度的缺陷,如:hp-5ms(30m
×
0.25mm
×
0.25μm)色谱柱,用于分析圆珠笔、中油笔等粘度较大的油墨中的溶剂成分如苯甲醇、苯氧乙醇、二甘醇乙醚等取得较好的效果;但发明人经过实验发现该色谱柱用于分析中性笔、水性笔等粘度较小的油墨中的强极性溶剂成分如乙二醇、1,2-丙二醇、1,3-丙二醇等会出现严重拖尾,并且各种油墨中广泛存在的丙三醇在该色谱柱上完全不成峰形;聚乙二醇(peg)为固定相的强极性柱(db-ffap柱(30m
×
0.25mm
×
0.25μm)、hp-innowax(30m
×
0.32mm
×
0.25μm)等)以10℃/min的升温速度进行气相色谱分析,丙三醇与三甘醇最大分离度也仅达到1.17,由于分离度过低(使用气相色谱仪分析需要化合物之间分离度达到1.5以上才能准确定量),无法实现准确定量。
[0052]
对于进样方式,本发明采用了不分流进样。由于书写墨迹中的溶剂成分属于痕量成分,尤其是在做书写时间的鉴定研究时,检测灵敏度越高对拐点的判断越准确。发明人研究发现,分流进样会在一定程度上会降低检测灵敏度,因而采用不分流进样的方式,从而实现提高灵敏度,降低检出限和定量限。
[0053]
可选地,所述柱温升温程序为:
[0054]
初始温度40~65℃保持0.5~3min,以40~130℃/min升温至110~135℃保持2~4min,以40~130℃/min升温至150~190℃保持1~4min,以40~130℃/min升温至220~260℃保持2~15min,后运行在240~260℃保持2~5min;
[0055]
或者:初始温度40~65℃保持0.5~3min,然后以40~130℃/min的速率升温到110~135℃并保持2~4min,然后以30~60℃/min的速率升温到220~260℃保持4~15min。
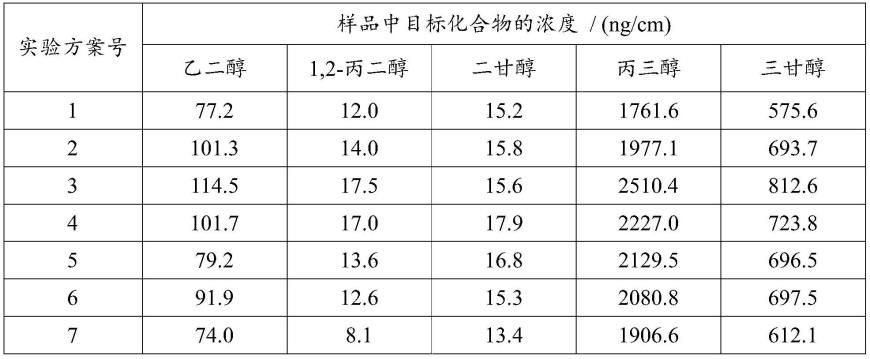
[0056]
可选地,所述柱温升温程序为:
[0057]
初始温度45~55℃保持0.5~1.5min,以80~120℃/min升温至120~130℃保持2~3min,以80~120℃/min升温至160~180℃保持1~2min,以80~130℃/min升温至220~
240℃保持2~4min,后运行在240~250℃保持2~3min;
[0058]
或者:初始温度45~55℃保持0.5~1.5min,然后以80~120℃/min的速率升温到120~130℃并保持2~4min,然后以35~45℃/min的速率升温到230~250℃保持4~10min。
[0059]
更优选地,步骤s3中,所述气相色谱分析条件为:
[0060]
柱流量:恒流模式2ml/min;
[0061]
进样口温度240℃,不分流进样,进样量1μl,吹扫时间0.5min,吹扫流量60ml/min;
[0062]
柱温升温程序:初始温度50℃保持1min,以100℃/min升温至125℃保持2min,以100℃/min升温至170℃保持1min,以100℃/min升温至230℃保持3min,后运行:以100℃/min升温至240℃保持3min;
[0063]
检测器温度:290℃。
[0064]
发明人研究发现,区别于传统升温程序以5~15℃/min较慢的匀速升温速率直接连接首尾两个等温保持阶段,上述柱温升温程序创新性地在50℃、230℃首尾两个等温保持阶段中间加入125℃、170℃两个等温保持阶段,各等温阶段之间以100℃/min的高速升温速率连接,这种创新在保证分离度的同时大大加快了分析速度,具有更优的检测效果。
[0065]
此外,在不分流进样的情况下,溶剂效应会使乙二醇、1,2-丙二醇、1,3-丙二醇等早流出的化合物峰展变宽、柱效降低。发明人研究发现,以50℃为柱温的初始温度,此时色谱峰的峰宽、对称因子、分离度、柱效、分析速度等参数综合考虑相对最优。
[0066]
优选地,所述气相色谱分析中,气相色谱仪为7890b型气相色谱仪(美国安捷伦公司),配备火焰离子化检测器(fid);数据采集、处理用openlab cds chemstation工作站完成。
[0067]
优选地,步骤s3中,所述定性检测的方法为:以样品溶液中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚的保留时间为定性依据,判断待测样品中目标化合物的存在。
[0068]
本方法中,采用目标化合物的保留时间与标准品的保留时间比对进行定性,若两者相对误差在
±
2%以内,则判定待测样品中含有该目标样品;也可以将标准品加入样品提取液中后进行色谱分析,通过观察各峰积分面积的增加情况来判断是否含有目标化合物。
[0069]
优选地,步骤s3中,所述定量检测的方法为:依据样品溶液中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚等各目标物分别与内标物的峰面积比值,根据各自的标准工作曲线计算待测样品中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚的含量。
[0070]
采用内标法进行定量分析,建立工作曲线后,由chemstation工作站积分计算出样品溶液中目标化合物的质量浓度ρi。待测样品中目标化合物的含量ci用单位长度的笔迹中所含目标化合物的质量来表示,计算方法如公式(1):
[0071][0072]
公式(1);
[0073]
式中ci为待测样品中目标化合物的含量,单位为ng/cm;
[0074]
ρi为测得样品溶液中目标化合物的质量浓度,单位为μg/ml;
[0075]
ρ0为测得空白纸张样品提取液中目标化合物的质量浓度,单位为μg/ml;
[0076]
v为样品溶液的体积,单位为μl;
[0077]
l为打孔取得笔迹的长度,单位为cm。
[0078]
同一份样品进样两次,计算结果取平均值,根据结果所处的浓度范围判断,其相对标准偏差在可接受范围内时结果有效。当结果低于检出限时,报告未检出(-);当结果高于检出限低于定量限时,报告检出未定量(nq);当结果高于定量限时,报告结果数值。
[0079]
与现有技术相比,本发明的有益效果是:
[0080]
本发明开发了一种墨迹中溶剂成分的检测方法。通过本发明的检测方法,合理选择内标物、制备样品溶液,并设定适宜的气相色谱分析条件,单个样品的检测耗时仅为12min左右,其中信号采集时间仅为8.8min左右,极大地缩短了检测耗时,大大提高了检测效率。此外,与常规的气相色谱-质谱联用法相比,本发明的方法仅采用气相色谱法进行检测分析,而无需联用质谱仪检测,在降低运行成本、减少仪器使用的情况下,实现了高灵敏度、高准确度、高精密度的检测效果,能够在一次检测过程中实现对9种墨迹种溶剂成分同时进行定性、定量分析。
附图说明
[0081]
图1为实施例1的取样方法示意图;
[0082]
图2为实施例3中使用hp-5ms(30m
×
0.25mm
×
0.25μm)色谱柱的气相色谱图;
[0083]
图3为实施例3中使用db-ffap柱(30m
×
0.25mm
×
0.25μm)色谱柱的气相色谱图;
[0084]
图4为实施例3中使用hp-innowax(30m
×
0.32mm
×
0.25μm)色谱柱的气相色谱图;
[0085]
图5为实施例3中使用db-alc2(30m
×
0.32mm
×
1.2μm)色谱柱的气相色谱图;
[0086]
图6为实施例4中1,2-丙二醇在不同初始温度(50℃、60℃、80℃)下的气相色谱图;
[0087]
图7为实施例6中n3号样品检测的气相色谱图;
[0088]
图8为实施例7中使用1,4-丁二醇为内标分离9种目标化合物的气相色谱图;
[0089]
图9为实施例8中使用3个等温阶段升温程序分离9种目标化合物的气相色谱图。
[0090]
在图2~图5、图7~图9中,色谱峰编号与化合物对应关系如下:1、乙二醇,2、1,2-丙二醇,3、1,3-丙二醇,4、1,3-丁二醇(内标),5、二甘醇乙醚,6、二甘醇,7、苯甲醇,8、丙三醇,9、苯氧乙醇,10、三甘醇,11、1,4-丁二醇(内标)。
具体实施方式
[0091]
为更好的说明本发明的目的、技术方案和优点,下面将结合具体实施例和附图来进一步说明本发明,但实施例并不对本发明做任何形式的限定。
[0092]
本发明的仪器与试剂的选取如下:
[0093]
气相色谱仪:7890b型气相色谱仪(美国安捷伦公司),配备火焰离子化检测器(fid);数据采集、处理用openlab cds chemstation工作站完成;
[0094]
色谱柱:db-alc2色谱柱(30m
×
0.32mm
×
1.2μm);
[0095]
分析天平:感量0.0001g;
[0096]
溶剂:甲醇(hplc纯);
[0097]
内标物:1,3-丁二醇(gc standard);
[0098]
目标化合物标准品:乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯
甲醇、2-苯氧乙醇、二甘醇乙醚(gc standard)
[0099]
除非特别说明,本发明采用的试剂、方法和设备为本技术领域常规试剂、方法和设备。除非特别说明,本发明所用试剂和材料均为市购。
[0100]
实施例1
[0101]
本实施例提供一种墨迹中溶剂成分的检测方法,包括如下步骤:
[0102]
s1.配制混合标准溶液:
[0103]
用分析天平精密称取1,3-丁二醇标准品,用甲醇配制浓度为10mg/ml的内标储备液,再用甲醇稀释定容,得到1,3-丁二醇浓度为50μg/ml的内标工作液;
[0104]
用分析天平精密分别称取目标化合物的标准品,用内标工作液配制成10mg/ml的混合标准溶液,然后用内标工作液稀释,配制成1、5、10、50、100、200、300μg/ml共7个级别含50μg/ml内标物的目标化合物的混合标准溶液。
[0105]
s2.制备样品溶液:
[0106]
用0.5mm打孔器在书写有墨迹的待测样品上打40个圆片,如图1所示,取笔迹样品总长度为2cm,放入0.2ml离心管,加入20μl内标工作液浸没混匀,将离心管在25℃水浴中静置40min,离心取上清液为样品溶液。
[0107]
s3.气相色谱分析:
[0108]
对所述混合标准溶液和样品溶液进行气相色谱分析,进行定性检测和定量检测;
[0109]
气相色谱分析条件为:
[0110]
色谱柱:db-alc2色谱柱,规格是30m
×
0.32mm
×
1.2μm;
[0111]
载气:氮气,纯度99.999%;
[0112]
柱流量:恒流模式2ml/min;
[0113]
进样口温度240℃,不分流进样,进样量1μl,吹扫时间0.5min,吹扫流量60ml/min;
[0114]
柱温升温程序:初始温度50℃保持1min,以100℃/min升温至125℃保持2min,以100℃/min升温至170℃保持1min,以100℃/min升温至230℃保持3min,后运行以100℃/min升温至240℃保持3min;
[0115]
检测器温度:290℃。
[0116]
步骤s3中,所述定性检测的方法为:以样品溶液中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚的保留时间为定性依据,判断待测样品中目标化合物的存在。
[0117]
本方法中,采用目标化合物的保留时间与标准品的保留时间比对进行定性,若两者相对误差在
±
2%以内,则判定待测样品中含有该目标样品;也可以将标准品加入样品提取液中后进行色谱分析,通过观察各峰积分面积的增加情况来判断是否含有目标化合物。
[0118]
步骤s3中,所述定量检测的方法为:依据样品溶液中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚等各目标物分别与内标物的峰面积比值,根据各自的标准工作曲线计算待测样品中乙二醇、1,2-丙二醇、1,3-丙二醇、二甘醇、三甘醇、丙三醇、苯甲醇、2-苯氧乙醇、二甘醇乙醚的含量。
[0119]
采用内标法进行定量分析,建立工作曲线后,由chemstation工作站积分计算出样品溶液中目标化合物的质量浓度ρi。待测样品中目标化合物的含量ci用单位长度的笔迹中所含目标化合物的质量来表示,计算方法如公式(1):
[0120][0121]
公式(1);
[0122]
式中ci为待测样品中目标化合物的含量,单位为ng/cm;
[0123]
ρi为测得样品溶液中目标化合物的质量浓度,单位为μg/ml;
[0124]
ρ0为测得空白纸张样品提取液中目标化合物的质量浓度,单位为μg/ml;
[0125]
v为样品溶液的体积,单位为μl;
[0126]
l为切取笔迹的长度,单位为cm,本实施例中l=2。
[0127]
同一份样品进样两次,计算结果取平均值,根据结果所处的浓度范围判断,其相对标准偏差在可接受范围内时结果有效。当结果低于检出限时,报告未检出(-);当结果高于检出限低于定量限时,报告检出未定量(nq);当结果高于定量限时,报告结果数值。样品中待测成分的含量与精密度可接受范围关系见《中华人民共和国药典(2020年版)》,四部:9101分析方法验证指导原则。
[0128]
实施例2
[0129]
为了研究提取条件对于提取效果的影响,本实施例以甲醇为提取剂,使用均匀设计法对提取的温度(10、20、25、30、35、40、50℃)和提取的时间(5、10、15、20、25、30、40min)组成的二因素七水平进行实验设计。采用均匀设计表,配合其使用表,组成七种实验方案,见表1。
[0130]
表1提取温度与时间因素的均匀设计方案
[0131]
实验方案号温度/(℃)时间/(min)1102522010325404302053556403075015
[0132]
取同一签字笔在同一时间制备的样品按照表1中的条件提取,除了步骤s2中提取方式变化之外,7个实验方案的其他步骤、条件均与实施例1相同。
[0133]
每种实验方案制备两个平行样品,每个样品重复测定两次,结果取平均值,实验结果见表2。由表中数据可知,采用3号实验方案(25℃,40min)提取样品,对大多数成分的提取率最高,故确定提取样品的优选条件为:以甲醇为提取剂,在25℃水浴中静置40min。
[0134]
表2不同提取温度和时间对样品中溶剂成分提取量的影响
[0135][0136]
实施例3
[0137]
为了研究色谱柱对于气相色谱分析效果的影响,本实施例采用hp-5ms(30m
×
0.25mm
×
0.25μm)色谱柱、db-ffap柱(30m
×
0.25mm
×
0.25μm)、hp-innowax(30m
×
0.32mm
×
0.25μm)、db-alc2色谱柱(30m
×
0.32mm
×
1.2μm)分别对样品溶液进行气相色谱分析;以9种目标物和内标物的混合标准溶液为测试样品,除了步骤s3的气相色谱分析条件外,其他步骤和条件均与实施例1相同。测试结果见图2~图5,各目标化合物与色谱峰的对照如下:1、乙二醇,2、1,2-丙二醇,3、1,3-丙二醇,4、1,3-丁二醇(内标),5、二甘醇乙醚,6、二甘醇,7、苯甲醇,8、丙三醇,9、苯氧乙醇,10、三甘醇。
[0138]
使用hp-5ms(30m
×
0.25mm
×
0.25μm)色谱柱的gc检测结果如图2所示,可以看出,中性笔、水性笔等粘度较小的油墨中的强极性溶剂成分乙二醇、1,2-丙二醇、1,3-丙二醇等会出现严重拖尾,并且各种油墨中广泛存在的丙三醇在该色谱柱上完全不成峰形。使用db-ffap柱(30m
×
0.25mm
×
0.25μm)色谱柱的gc检测结果如图3所示,使用hp-innowax(30m
×
0.32mm
×
0.25μm)色谱柱的gc检测结果如图4所示,可以看出,这两种色谱柱对于丙三醇与三甘醇的分离度很差,分离效果稍好的hp-innowax仅达到1.17,而使用气相色谱仪分析需要目标化合物之间分离度达到1.5以上才能准确定量,并且签字笔油墨中经常同时含有丙三醇与三甘醇,因此上述聚乙二醇类色谱柱不适用于气相色谱法定量分析书写墨迹中的溶剂成分。使用db-alc2色谱柱(30m
×
0.32mm
×
1.2μm)的gc检测结果如图5所示。在本发明的气相色谱分析条件下,db-alc2色谱柱能将9种目标化合物与内标完全分离,且柱效高、峰形尖锐对称,包括后运行的整个气相循环时间仅需12min。由图5与图3、图4比较可见,本方法使用的db-alc2色谱柱,所有目标物化合物在7.5min出峰完毕,而此时传统方法使用的色谱柱还没有开始出峰。
[0139]
使用db-alc2色谱柱分离9种目标物及内标物的保留时间见表3。
[0140]
表3 9种目标化合物及内标物在db-alc2色谱柱上的保留时间
[0141]
色谱峰编号目标化合物保留时间/(min)1乙二醇3.90921,2-丙二醇4.10131,3-丙二醇4.80141,3-丁二醇4.9865二甘醇乙醚5.440
6二甘醇5.7567苯甲醇5.9358丙三醇6.19692-苯氧乙醇6.99010三甘醇7.249
[0142]
实施例4
[0143]
为了研究柱温的初始温度对于气相色谱分析效果的影响,本实施例研究了在不分流进样情况下,1,2-丙二醇在不同初始温度(45、50、55、60、65、80℃)下的峰形,其中50℃、60℃、80℃时的峰形如图6所示。
[0144]
研究发现,初始柱温在50℃以下时色谱峰的峰宽、对称因子、分离度、柱效等参数良好,在55℃以上时色谱峰各参数开始变差,在80℃时峰宽很宽,拖尾严重,影响定量,因此选择50℃为升温程序的优选初始温度。
[0145]
实施例5
[0146]
本实施例提供上述墨迹中溶剂成分的检测方法的方法学验证。
[0147]
(1)工作曲线的建立与检测限、定量限的测定
[0148]
按实施例1的方法配制混合标准溶液并进行气相色谱分析,用内标法进行定量计算。用最小二乘法以各目标化合物与内标物的峰面积比(y)对各目标化合物与内标物的浓度比(x)进行线性回归分析,得到各个目标化合物的标准工作曲线的线性方程及相关系数。
[0149]
将1μg/ml的混合标准溶液加入空白样品处理进样12次,计算各目标化合物测定结果的标准偏差,按下列公式(2)计算检出限和定量限,再用公式(1)换算成待测样品的检出限和定量限;
[0150][0151]
式(2);
[0152]
式中c
l
为检出限/定量限;
[0153]
k为置信因子,计算检出限时取3,计算定量限时取10;
[0154]
si为样品测定结果的标准偏差;
[0155]
c为待测目标化合物的理论含量;
[0156]
为样品测定结果的平均值。
[0157]
分析结果见表4,可见9种目标化合物各自的工作曲线在1~300μg/ml浓度范围内皮尔逊相关系数r均大于0.999,线性关系良好,换算成样品含量的线性范围为10~3000ng/cm;样品的检出限为0.3~4.7ng/cm,定量限为1.0~14.1ng/cm。
[0158]
表4
[0159][0160][0161]
(2)准确度和精密度
[0162]
采用空白加标法来计算加标回收率。按前述样品制备方法用空白纸张制取空白样品,按低、中、高三种浓度水平加入9种目标分析物标准品并进行色谱分析,每个水平重复测定6次。由空白样品加标量、加标后测得量分别计算9种目标化合物的回收率,结果见表5。可见9种目标物的加标回收率在98.1%~105.1%之间,相对标准偏差在0.2%~2.1%,各水平添加样品的回收率在其所属的浓度区间均符合要求,说明方法准确度高,重复性良好。
[0163]
表5
[0164][0165][0166]
实施例6
[0167]
本实施例为对30种市售中性笔、水性笔、圆珠笔的墨迹中溶剂成分的检测。采用实施例1的方法,对市场上购买的10种中性笔(分别记为n1~n10)、10种水性笔(直液式走珠笔)(分别记为w1~w10)、10种圆珠笔(分别记为b1~b10)进行了分析,30种笔样品均为不同牌号的产品,检测结果见表6。其中n3号样品检测的色谱图见图7。
[0168]
表6
[0169][0170][0171]
*.-,未检出;nq,检出未定量;样品在测定前先稀释一倍,以使丙三醇含量在线性范围。
[0172]
实施例7
[0173]
本实施例提供一种墨迹中溶剂成分的检测方法,与实施例1的区别在于:
[0174]
步骤s1中,使用的内标物为1,4-丁二醇。
[0175]
按照此方法,9种目标化合物的色谱分离效果见图8,由图可见1,4-丁二醇的峰在二甘醇乙醚与二甘醇之间。1,4-丁二醇与两者的分离度分别为5.00和5.99。而使用1,3-丁二醇为内标时二甘醇乙醚与二甘醇之间没有内标物,两者分离度达到10.79。由此可知,以1,4-丁二醇为内标物会使二甘醇乙醚与二甘醇的分离度变小。
[0176]
因此在本发明的气相色谱分析条件下,采用1,4-丁二醇为内标物在绝大多数情况下能取得良好的效果,但在二甘醇乙醚与二甘醇的分离度指标方面不如以1,3-丁二醇作为内标物的实施例。
[0177]
实施例8
[0178]
本实施例提供一种墨迹中溶剂成分的检测方法,与实施例1的区别在于:
[0179]
步骤s3中,气相色谱分析条件为:
[0180]
柱温升温程序:初始温度45℃保持1min,然后以100℃/min的速率升温到125℃并保持2.5min,然后以40℃/min的速率升温到230℃保持3min,后运行:以100℃/min升温至240℃保持3min。
[0181]
按照此方法,各目标化合物的色谱分离效果见图9。由图可知各目标化合物分离效果良好,但所需时间比实施例1稍长。本发明实施例1中各等温阶段的温度调整均需相应调整各阶段的保持时间,可见在本实施例中去掉170℃的等温阶段对整个分析过程的效率略有不利影响。
[0182]
对比例1
[0183]
本对比例提供一种墨迹中溶剂成分的检测方法,与实施例1的区别在于:
[0184]
步骤s3中,气相色谱分析条件为:
[0185]
柱温升温程序:初始温度50℃保持1min,以10℃/min升温至230℃保持5min。
[0186]
按照此方法,虽然各目标化合物能够完全分离,但由于柱温升温程序采用的是传统的匀速升温方法,整个气相循环时间为24min,耗时远比本发明实施例1中的12min要久。
[0187]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1