一种包含大黄和附子的中药复方的指纹图谱构建方法及其应用与流程
本发明涉及医药,具体涉及一种包含大黄和附子的中药复方的指纹图谱构建方法及其应用。
背景技术:
1、
2、中药经典名方汤剂作为传统的中医临床用药的最常用的剂型,具有组方合理、起效迅速、疗效显著、易于吸收等优点,深受广大患者的信赖。却因调配、携带、临时煎煮、久置容易发生霉败变质、汤液味苦和量大,以及标准难以统一,严重影响其临床疗效等缺点,不能适应现代人的生活要求。为了保持汤剂的优势,克服汤剂的种种不足,国家药品监督管理局药品审评中心于2021年08月31日发布的《按古代经典名方目录管理的中药复方制剂药学研究技术指导原则(试行)》中明确指出按古代经典名方目录管理的中药复方制剂的质量应与经典名方基准样品的质量基本一致。基准样品代表了制剂的整体内在质量,除了成型工艺外,基准样品与制剂的其余质量控制指标均基本一致。因此基准样品是制剂内在质量的实物对照,是大生产工艺优化及其质量标准制定的参照物。基准样品是经典名方乃至所有中药研发的基准,是保证药物的安全性和有效性的对照物。经典名方复方制剂生产工艺路线制定、参数的优化和质量标准制定要以经典名方基准样品为参照。基准样品是沟通临床-企业-科研的纽带,是传承、应用和发扬传统中医药的基准。
3、指纹图谱是基于中药物质群整体作用的认识,借助于波谱和色谱技术获得中药化学成分的光谱或色谱图,是实现鉴别中药真实性、评价质量一致性和产品稳定性的可行模式,具有信息量大、特征性强、整体性和模糊性等特点。中药指纹图谱能较全面地反映药材所含化学成分的相对关系,体现了中药成分的复杂性和相关性,与中医药的传统理论相适应,能真正对中药内在质量进行有效表征、综合评价和全面控制,尤其适用于有效成分不完全明确或不需要完全明确的情况下,对中药材及中药产品进行质量控制。中药指纹图谱除能用于考察中药材产地、采收季节、采收部位、炮制加工、储存时间等因素,以提供依据鉴别生产前原料药材的真伪优劣,更可用于中药生产过程的质量控制:追踪制剂中某些化学成分的变化,监测原料药材与成品之间、成品的各批次间质量的一致性及稳定性。与指标成分含量测定的质量分析方法相比,指纹图谱能够比较全面地反映中药化学成分的种类和数量,在中药复方制剂有效成分尚未完全阐明的现状下,可实现对中药内在质量的综合评价和对其整体物质的有效控制,是目前中药及其制剂质量控制的有效手段之一。
4、目前,尚未见文献报道对本发明所要求保护的包含大黄和附子的中药复方的化学成分进行全面地分析以及指纹图谱研究,现有技术中,仅对该中药复方中的单一成分进行质量分析,尚无较全面、系统的质量控制方法反映本发明的中药复方及成品中主要基准样品成分的质量状况,无法对其生产过程及产品质量有效控制,不能较好地保证其临床疗效,故需采用能全面控制本发明的中药复方整体质量的指纹图谱的质控方法控制其关键质量。
技术实现思路
1、基于此,本发明提供了一种包含大黄和附子的中药复方的检测方法,其包括如下步骤:
2、取中药复方供试品溶液和对照品溶液进行检测,其中,该中药复方包括大黄、人参、甘草、干姜和附子,该对照品为甘草素、异甘草素、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、6-姜辣素和甘草次酸,
3、该检测的色谱条件为:采用填料为t3的色谱柱,流动相a选自乙腈、甲醇和四氢呋喃中的一种或多种,流动相b为酸水溶液、碱水溶液和/或缓冲盐水溶液,梯度洗脱程序为:0~1min,30%~32%a;1~8min,32%~63%a;8~13min,63%~67%a;13~14min,67%~95%a,流速为0.4~1.5ml/min,柱温为30~50℃,检测波长为200~400nm;
4、根据检测结果,获得该中药复方的成分信息,或者成分信息和含量信息。
5、进一步地,该信息为根据所记录的该中药复方供试品溶液的色谱图和该对照品溶液的色谱图中的相应峰面积,按照外标法计算该中药复方中以下成分中一种或多种的含量:甘草素、异甘草素、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、6-姜辣素和甘草次酸。
6、进一步地,该中药复方供试品溶液的制备方法包括以下步骤:称取适量的中药复方粉末,添加水或醇,经过振摇提取、超声提取和/或回流提取之后,放冷、补足重量、摇匀、过滤,取第一续滤液通过固相萃取小柱,使用醇进行洗脱,收集流出液和洗脱液,浓缩至接近干燥,加酸进行酸水解,有机溶剂萃取之后合并有机溶剂层,回收溶剂至干燥,残渣加醇溶解,过滤,取第二续滤液,得到该中药复方供试品溶液。
7、进一步地,该中药复方粉末的制备方法包括以下步骤:称取适量的大黄、人参、甘草、干姜和附子,得到饮片混合物,加水浸泡一段时间之后,武火加热至沸腾后文火煎煮一段时间,过滤,滤液浓缩成流膏,取出,干燥,研磨成粉,得到该中药复方粉末。
8、进一步地,该饮片混合物与该水的质量/体积之比为1:4~50,例如约1:44。
9、进一步地,该加水浸泡的时间为0.2~1h,例如约0.5h。
10、进一步地,该文火煎煮的时间为0.5~1.5h,例如约1h。
11、进一步地,该过滤为100~300目尼龙滤布过滤。
12、进一步地,该浓缩为减压浓缩或蒸干浓缩。
13、进一步地,该流膏为在20℃下相对密度为1.10~1.15的流膏。
14、进一步地,该干燥为真空干燥或冷冻干燥。
15、进一步地,该对照品溶液的制备方法包括:称取适量的甘草素、异甘草素、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、6-姜辣素和甘草次酸;以及添加体积百分比浓度为10%~100%的甲醇水溶液例如约75%的甲醇水溶液制成含各成分浓度为1~200μg/ml的该对照品溶液。
16、进一步地,该高效液相检测的该流速为0.6~1.2ml/min,例如约0.9ml/min。
17、进一步地,该柱温为35~45℃,例如约40℃。
18、进一步地,该检测波长为250~320nm,例如约280nm。
19、进一步地,该进样量为5~20μl,例如8~15μl,例如约10μl。
20、进一步地,该大黄酚对应的色谱峰的理论塔板数不低于10000。
21、进一步地,该对照品对应的色谱峰的分离度大于2.0。
22、进一步地,该色谱柱为cortecs t3色谱柱。
23、进一步地,该cortecs t3色谱柱为cortecs t3色谱柱,3.0mm×150mm,2.7μm。
24、进一步地,该检测方法进一步包括缺各单味饮片阴性对照溶液的制备与检测:该缺各单味饮片的中药复方阴性对照溶液的制备方法与该中药复方供试品溶液的制备方法相同。
25、进一步地,在该中药复方供试品溶液的制备方法中,该醇为甲醇。
26、进一步地,该甲醇的体积浓度百分比为50%~100%,例如约50%或约75%。
27、进一步地,该超声提取的功率为300~700w,例如约500w。
28、进一步地,该超声提取的频率为30~50khz,例如约40khz。
29、进一步地,该中药复方与该醇的质量/体积(g/ml)之比为1:20~1:60,例如约1:25或约1:50。
30、进一步地,该固相萃取小柱为pep或c18固相萃取小柱。
31、进一步地,该固相萃取小柱的规格为200mg、300mg或500mg。
32、进一步地,该浓缩为减压浓缩或蒸干浓缩。
33、进一步地,该酸为盐酸和/或硫酸。
34、进一步地,该酸水解为先加入酸进行超声处理,再加入有机溶剂进行两相加热回流酸水解。
35、进一步地,该有机溶剂为乙酸乙酯、三氯甲烷和/或乙醚。
36、进一步地,该萃取为振摇萃取。
37、进一步地,该干燥为真空干燥或冷冻干燥。
38、进一步地,该过滤是采用微孔滤膜过滤。
39、进一步地,该流动相a为甲醇-乙腈(28:72)。
40、进一步地,该酸水溶液、碱水溶液和/或缓冲盐水溶液选自不同浓度的弱酸及其盐、弱碱及其盐中的一种或多种。
41、进一步地,该酸水溶液、碱水溶液和/或缓冲盐水溶液选自不同浓度的甲酸、冰乙酸、磷酸、三氟乙酸、甲酸和甲酸铵、乙酸和乙酸钠、乙酸和乙酸铵、磷酸氢二钠和磷酸二氢钠、磷酸氢二钠和磷酸二氢钾、磷酸氢二钠和柠檬酸、柠檬酸和柠檬酸钠、甘氨酸和盐酸、或者邻苯二甲酸和盐酸。
42、进一步地,该酸水溶液为0.01%~0.1%的酸水溶液。
43、进一步地,该酸水溶液为0.01%~0.1%的三氟乙酸水溶液。
44、进一步地,该酸水溶液为约0.03%的三氟乙酸水溶液。
45、进一步地,该缓冲盐水溶液为磷酸盐水溶液和/或醋酸盐水溶液。
46、进一步地,该缓冲盐水溶液的ph值不大于7.0。
47、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为(0.5~5):(0.5~5):(0.5~5):(0.5~5):(0.5~5)。
48、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为(0.5~2.5):(0.5~2.5):(0.5~2.5):(0.5~2.5):(0.5~2.5)。
49、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为约2:约1:约1:约1:约1。
50、进一步地,该对照品溶液为甘草素浓度为60~70μg/ml、异甘草素浓度为5~10μg/ml、芦荟大黄素浓度为5~10μg/ml、大黄酸浓度为0.5~5μg/ml、大黄素浓度为5~10μg/ml、大黄酚浓度为10~20μg/ml、大黄素甲醚浓度为0.5~5μg/ml、6-姜辣素浓度为1~10μg/ml和甘草次酸浓度为40~100μg/ml的混合溶液。
51、进一步地,该对照品溶液为甘草素浓度为约66μg/ml、异甘草素浓度为约7μg/ml、芦荟大黄素浓度为约7μg/ml、大黄酸浓度为约2μg/ml、大黄素浓度为约6μg/ml、大黄酚浓度为约12μg/ml、大黄素甲醚浓度为约2μg/ml、6-姜辣素浓度为约4μg/ml和甘草次酸浓度为约51μg/ml的混合溶液。
52、根据本发明的另一个方面,提供了一种包含大黄和附子的中药复方的指纹图谱构建方法,其包括如下步骤:
53、中药复方供试品溶液的制备:称取适量的中药复方粉末,添加水或醇,经过振摇提取、超声提取和/或回流提取之后,放冷、补足重量、摇匀、过滤,取第一续滤液通过固相萃取小柱,使用醇进行洗脱,收集流出液和洗脱液,浓缩至接近干燥,加酸进行酸水解,有机溶剂萃取之后合并有机溶剂层,回收溶剂至干燥,残渣加醇溶解,过滤,取第二续滤液,得到该中药复方供试品溶液,其中,该中药复方包括大黄、人参、甘草、干姜和附子;
54、对照品溶液的制备:称取适量的甘草素、异甘草素、芦荟大黄素、大黄酸、大黄素、大黄酚、大黄素甲醚、6-姜辣素和甘草次酸;以及添加体积百分比浓度为10%~100%的甲醇水溶液例如约75%的甲醇水溶液制成含各成分浓度为1~200μg/ml的该对照品溶液;
55、根据高效液相检测该中药复方供试品溶液和该对照品溶液的结果,获得中药复方指纹图谱;
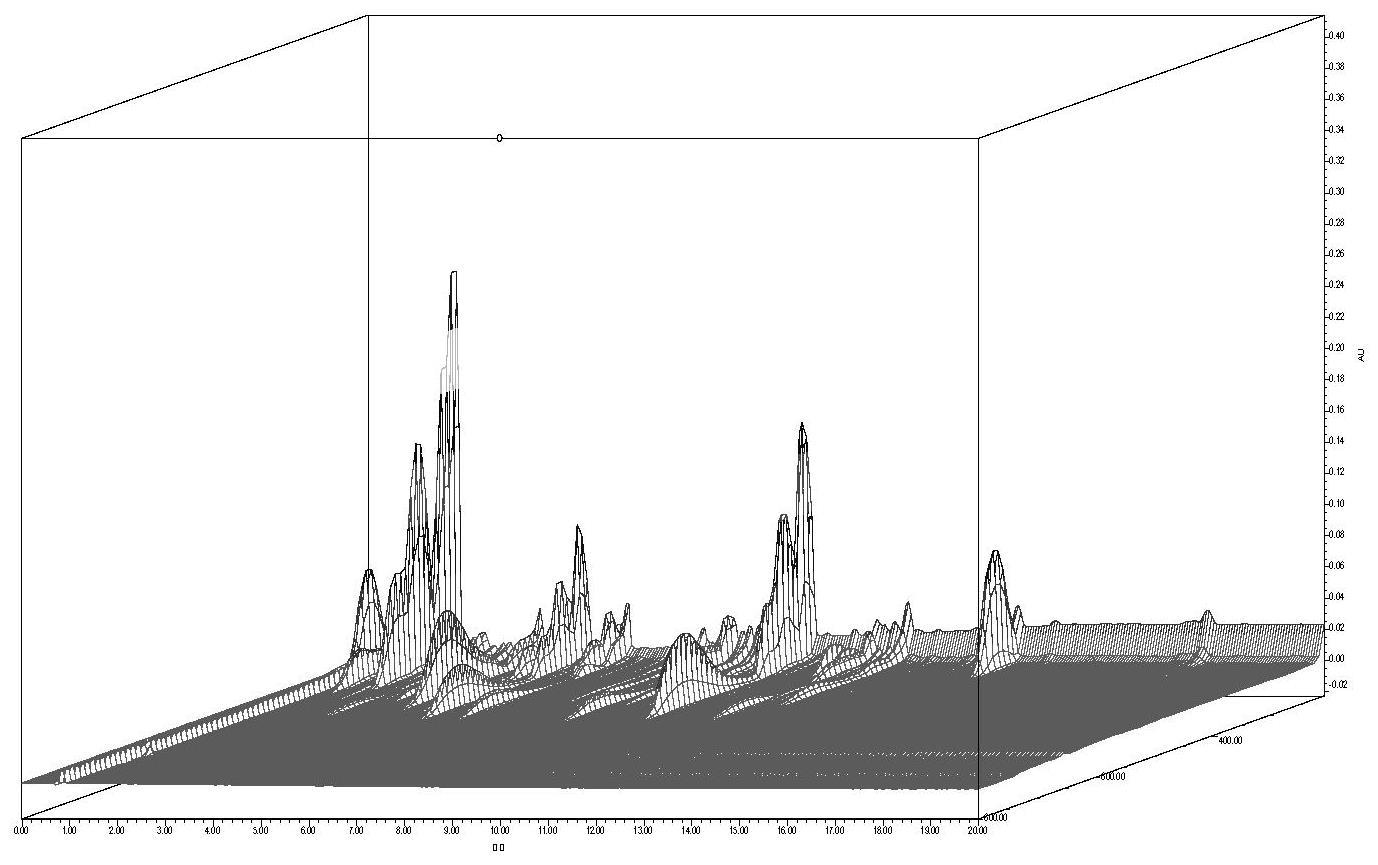
56、该高效液相检测的色谱条件为:采用填料为t3的色谱柱,流动相a选自乙腈、甲醇和四氢呋喃中的一种或多种,流动相b为酸水溶液、碱水溶液和/或缓冲盐水溶液,梯度洗脱程序为:0~1min,30%~32%a;1~8min,32%~63%a;8~13min,63%~67%a;13~14min,67%~95%a,流速为0.4~1.5ml/min,柱温为30~50℃,检测波长为200~400nm。
57、进一步地,该高效液相检测的该流速为0.6~1.2ml/min,例如约0.9ml/min。
58、进一步地,该柱温为35~45℃,例如约40℃。
59、进一步地,该检测波长为250~320nm,例如约280nm。
60、进一步地,该进样量为5~20μl,例如8~15μl,例如约10μl。
61、进一步地,该大黄酚对应的色谱峰的理论塔板数不低于10000。
62、进一步地,该对照品对应的色谱峰的分离度大于2.0。
63、进一步地,在该检测波长为280nm时,该指纹图谱包括1-12号峰,其中,9号峰为大黄酚作为参照峰,1号峰为甘草素,3号峰为异甘草素,5号峰为芦荟大黄素,6号峰为大黄酸,7号峰为6-姜辣素,8号为大黄素,10号为大黄素甲醚,12号峰为甘草次酸,其保留时间分别对应为10.0~10.2min、2.5~2.7min、4.9~5.1min、5.8~6.0min、6.4~6.6min、6.6~6.8min、8.5~8.7min、11.5~11.7min、14.8~15.0min。
64、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为(0.5~5):(0.5~5):(0.5~5):(0.5~5):(0.5~5)。
65、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为(0.5~2.5):(0.5~2.5):(0.5~2.5):(0.5~2.5):(0.5~2.5)。
66、进一步地,该中药复方中大黄、人参、甘草、干姜和附子的重量比为约2:约1:约1:约1:约1。
67、进一步地,该对照品溶液为甘草素浓度为60~70μg/ml、异甘草素浓度为5~10μg/ml、芦荟大黄素浓度为5~10μg/ml、大黄酸浓度为0.5~5μg/ml、大黄素浓度为5~10μg/ml、大黄酚浓度为10~20μg/ml、大黄素甲醚浓度为0.5~5μg/ml、6-姜辣素浓度为1~10μg/ml和甘草次酸浓度为40~100μg/ml的混合溶液。
68、进一步地,该对照品溶液为甘草素浓度为约66μg/ml、异甘草素浓度为约7μg/ml、芦荟大黄素浓度为约7μg/ml、大黄酸浓度为约2μg/ml、大黄素浓度为约6μg/ml、大黄酚浓度为约12μg/ml、大黄素甲醚浓度为约2μg/ml、6-姜辣素浓度为约4μg/ml和甘草次酸浓度为约51μg/ml的混合溶液。
69、进一步地,该流动相a为甲醇-乙腈(28:72)。
70、进一步地,该酸水溶液、碱水溶液和/或缓冲盐水溶液选自不同浓度的弱酸及其盐、弱碱及其盐中的一种或多种。
71、进一步地,该酸水溶液、碱水溶液和/或缓冲盐水溶液选自不同浓度的甲酸、冰乙酸、磷酸、三氟乙酸、甲酸和甲酸铵、乙酸和乙酸钠、乙酸和乙酸铵、磷酸氢二钠和磷酸二氢钠、磷酸氢二钠和磷酸二氢钾、磷酸氢二钠和柠檬酸、柠檬酸和柠檬酸钠、甘氨酸和盐酸、或者邻苯二甲酸和盐酸。
72、进一步地,该酸水溶液为0.01%~0.1%的酸水溶液。
73、进一步地,该酸水溶液为0.01%~0.1%的三氟乙酸水溶液。
74、进一步地,该酸水溶液为约0.03%的三氟乙酸水溶液。
75、进一步地,该缓冲盐水溶液为磷酸盐水溶液和/或醋酸盐水溶液。
76、进一步地,该缓冲盐水溶液的ph值不大于7.0。
77、进一步地,该色谱柱为cortecs t3色谱柱。
78、进一步地,该cortecs t3色谱柱为cortecs t3色谱柱,3.0mm×150mm,2.7μm。
79、进一步地,在该检测波长为280nm时,1号峰、3号峰和12号峰来自药材甘草,2号峰、4号峰、5号峰、6号峰、8号峰、9号峰、10号峰和11号峰来自药材大黄,7号峰来自药材干姜。
80、进一步地,在该检测波长为280nm时,包括12个共有峰,以大黄酚9号色谱峰为参比峰,其他11个共有峰的相对保留时间的rsd%依次为1号色谱峰0.59%、2号色谱峰0.19%、3号色谱峰0.32%、4号色谱峰0.47%、5号色谱峰0.05%、6号色谱峰0.21%、7号色谱峰0.42%、8号色谱峰0.06%、10号色谱峰0.16%、11号色谱峰0.17%和12号色谱峰0.29%。
81、根据本发明的另一个方面,提供了一种包含大黄和附子的中药复方的质量控制方法,该方法包括如下步骤:
82、(1)根据上述指纹图谱构建方法建立中药复方基准样品标准指纹图谱;
83、(2)取中药复方供试品溶液,根据上述指纹图谱构建方法中的色谱条件进行检测,得到中药复方待测样品指纹图谱;以及
84、(3)将步骤(2)所得到的该中药复方待测样品指纹图谱与步骤(1)所得到的该中药复方基准样品标准指纹图谱进行对比,符合则为合格产品,不符合则为不合格产品。
85、进一步地,该符合的要求包括以下的一种或多种:
86、(1)该中药复方待测样品指纹图谱中呈现出12个特征色谱峰,其中9个色谱峰分别与该中药复方基准样品标准指纹图谱中相应的对照品色谱峰保留时间值的±10%之内;
87、(2)以大黄酚对照品所对应的峰为s峰,中药复方待测样品指纹图谱中的各特征色谱峰与s峰的相对保留时间在该中药复方基准样品标准指纹图谱的各特征色谱峰的相对保留时间值的±10%之内;
88、(3)按中药色谱指纹图谱相似度评价系统,该中药复方待测样品指纹图谱与该中药复方基准样品标准指纹图谱经相似度计算,相似度不得低于0.90。
89、根据本发明的另一个方面,提供了一种上述检测方法或上述指纹图谱构建方法或上述质量控制方法在包含大黄和附子的中药复方的质量检测和/或质量评价和/或质量控制中的用途。
90、本发明的有益效果:
91、简而言之,本发明提供了一种包含大黄和附子的中药复方基准样品的指纹图谱测定和质量控制方法,该方法确认了12个共有特征峰,条件简单,分析时间短,解决了指纹特征峰难以分开和杂质峰的干扰问题,保证了基准样品的化学组成稳定性和使用安全性,为后续制剂的质量控制提供了重要的参考依据,确保产品的质量稳定,保障中药复方的疗效,让该中药复方更好地为人类生命健康服务。
92、具体而言,与现有技术相比,本发明的有益效果如下:
93、(1)建立本发明中药复方基准样品的指纹图谱,克服了单一成分含量测定难以反映整体含量的缺陷,可以从整体上、宏观上控制本发明中药复方基准样品的内在质量,保证了药物的疗效,利用现代药物研究的主要手段,实现精品传承经典,使经典名方得到了更为正规的质量控制。
94、(2)本发明中药复方基准样品中化学成分复杂,要实现其特征峰的分离难度大,本发明在建立起指纹图谱的过程中,采用了梯度洗脱的方法,解决了指纹特征峰难以分开和杂质峰的干扰问题。
95、(3)本发明在建立本发明中药复方基准样品的指纹图谱过程中,确认了12个共有特征峰,并对其相对保留时间和相对峰面积和相似度进行了研究,保证了基准样品的化学组成稳定性和使用安全性,为后续真武汤复方制剂的质量控制提供重要的参考依据和质量参照。
96、(4)以本发明中药复方基准样品中各有效成分指纹图谱作为一个整体看待,注重各个特征峰的前后顺序和相互关系,既避免了因只测定一、二个化学成分而判定本发明中药复方基准样品整体质量的片面性,又减少了为质量达标而人为处理的可能性,为完整、准确评价本发明中药复方基准样品的质量提供了新的方法和手段。
97、(5)本发明方法稳定性好、精密度高、重现性好、便捷且易于掌握。
- 还没有人留言评论。精彩留言会获得点赞!