性状预测模型制作方法和性状预测方法与流程
本发明涉及一种性状预测模型制作方法和性状预测方法。
背景技术:
目前,作为使用人基因组信息的表现型预测,着眼于性状感受性多态性,集中研究了仅使用已经被鉴定的感受性多态性预测表现型的方法(v.lyssenkoetal.,nengljmed2008vol.359p.2220-2232;s.ripatthietal.,lanet2010vol.376p.1393-1400;c.a.ibrahim-verbaasetal.,stroke2014vol.45p.403-412)。这些方法具体列举与性状相关的数个到数百个多态性,推定各个多态性的重要性,能够将各个多态性对性状的影响数值化,从这点上说,容易直观地理解。
但是,仅使用感受性多态性这一点是该方法的缺点,是有限度的。这是因为在几乎全部的多因子性状中,在成为实际的原因的感受性多态性中被鉴定的感受性多态性极少。例如,推定能够由遗传因素说明身高的方差中的约80%,但是能够由已知的感受性多态性来说明的方差只不过为5%左右。
因此,非专利文献(d.speedandd.j.balding,genomeresearch2015vol.24p.1550-1557)中记载了不管是不是感受性多态性都使用网罗性(全基因组)的多态性信息的表现型预测法。即,将多个单核苷酸多态性(singlenucleotidepolymorphism;snp)分解为多个类别(category),应用线性混合模型。但是,在该方法中,预测的精度也还不充分。
技术实现要素:
发明所要解决的课题
本发明的目的在于提供一种用于从单核苷酸多态性数据预测性状的表现型的性状预测模型制作方法和能够以高的准确率预测性状的性状预测方法。
用于解决课题的方法
本发明的发明人研究了不管是否为感受性多态性都使用网罗性(全基因组)的多态性信息的统计学处理方法。即,发现:以包含身高或hbalc检查值的27个量的性状、和包含患有糖尿病或低hdl胆固醇血症的5个质的性状为例,将约100万个多态性作为基因组信息,将性别年龄信息用作调节变量,应用线性混合模型,对性状进行学习而制作预测模型,其结果,该预测与实测值存在高度地相关。这样,我们完成了从基因组信息预测表现型的预测方法。
本发明的一个实施方式为一种性状预测模型制作方法,其使用在生物的多个个体中使多个单核苷酸多态性与形状对应的单核苷酸多态性数据,制作预测多因子性性状的表现型的预测模型,该性状预测模型制作方法包括:将上述多个单核苷酸多态性分别矩阵表示的工序;将上述多个单核苷酸多态性基于遗传结构分类为多个类别的工序;对于各个上述类别,使用上述矩阵表示和属于上述类别的上述单核苷酸多态性的个数计算基因组相似度矩阵的工序;和将上述基因组相似度矩阵和上述遗传结构的参数应用于线性混合模型的工序。上述遗传结构可以为效应量(effectsize)和/或等位基因频率(allelefrequency)。
本发明的另一实施方式为一种性状预测模型制作方法,其使用在生物的多个个体中使多个单核苷酸多态性、性别、年龄与形状对应的单核苷酸多态性数据,制作预测多因子性性状的表现型的性状预测模型,该性状预测模型制作方法包括:将上述多个单核苷酸多态性分别矩阵表示的工序;将上述性别和/或年龄矩阵表示的工序;使用上述单核苷酸多态性的矩阵表示和上述单核苷酸多态性的个数计算基因组相似度矩阵的工序;和将上述基因组相似度矩阵、和上述性别和/或年龄的矩阵应用于线性混合模型的工序。上述性状可以选自身高、体重、最高血压、最低血压、血糖、hbalc、红细胞数、血色素、血细胞容积、白细胞数、血小板数、嗜中性粒细胞的比例、淋巴细胞的比例、单核细胞的比例、嗜酸性粒细胞的比例、嗜碱性粒细胞的比例、大型不染色细胞的比例、ast(got)、alt(gpt)、γ-gtp、总胆固醇、中性脂肪、hdl胆固醇、ldl胆固醇、肌酐、尿素氮、尿酸、糖尿病、高血压症、高ldl胆固醇血症、低hdl胆固醇血症、高甘油三酯血症。
本发明的再一实施方式为一种性状预测方法,其在生物个体中从多个单核苷酸多态性数据预测该生物个体的性状,该性状预测方法包括:使用学习用数据集,按照上述预测模型制作方法制作预测模型的工序;确定线性混合模型的参数和潜在变量的工序;和将该生物个体的上述多个单核苷酸多态性数据应用于上述预测模型的工序。
本发明的再一实施方式为一种用于在生物个体中从多个单核苷酸多态性数据预测该生物个体的性状的程序,其中,使计算机执行上述性状预测方法。本发明的一个实施方式可以为一种存储有本程序的计算机可读取的存储介质。
本发明的再一实施方式为一种用于在生物个体中从多个单核苷酸多态性数据预测该生物个体的性状的性状预测系统,其具备:
(i)用于输入上述生物个体的多个单核苷酸多态性数据的输入装置;
(ii)使用输入的数据,执行上述程序的计算机;和
(iii)用于输出由(ii)得到的结果的输出装置。
==与关联文献的交叉引用==
本申请主张基于在2014年11月25日申请的日本国专利申请2014-238252的优先权,通过引用该基础申请,包含在本说明书中。
附图说明
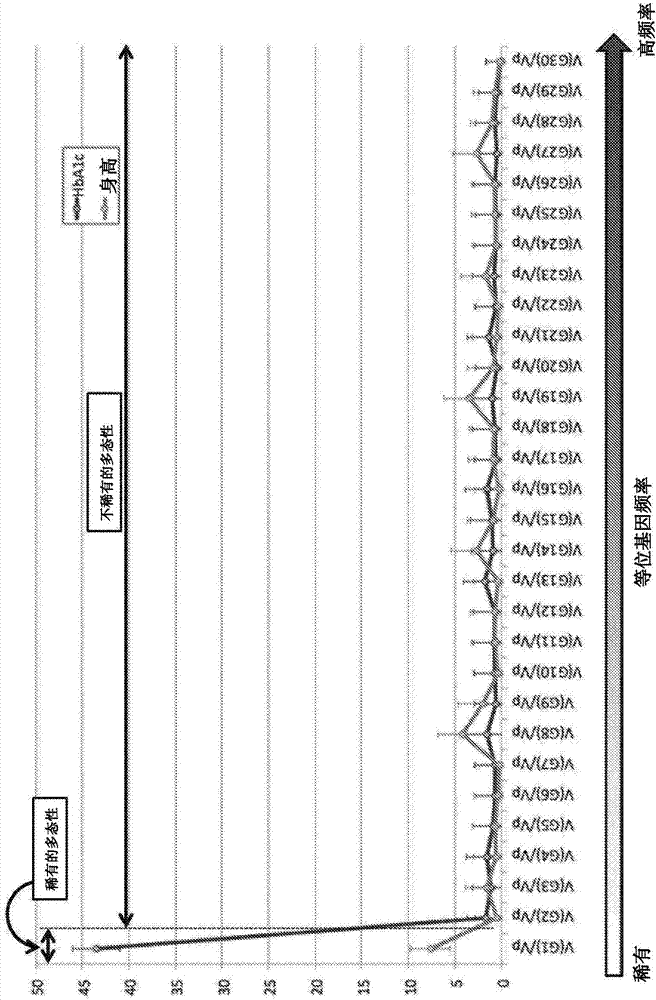
图1是表示在本发明的一个实施例中,着眼于hbalc检查值和身高,利用遗传结构分解法的贡献率的推定结果的图(qes=50、qraf=1的情况)。
图2是表示在本发明的一个实施例中,着眼于hbalc检查值和身高,利用遗传结构分解法的贡献率的推定结果的图(qes=1、qraf=30的情况)。
图3是在本发明的一个实施例中实施例中所使用的性状的一览。
图4是表示在本发明的一个实施例中27个量的性状的精度评价结果的图。对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)这3种方法进行比较。作为评价指标,使用实测值和预测值的r2(相关系数的平方),利用二折交叉验证(2-foldcrossvalidation)法进行评价。
图5是表示在本发明的一个实施例中5个质的性状的精度评价结果的图。对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)这3种方法进行比较。作为评价指标,使用auc,利用二折交叉验证法进行评价。
图6是表示在本发明的一个实施例中样本量充分大时27个量的性状的精度评价结果的图。对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)、(4)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=10、qraf=1的情况(有遗传结构分解;本发明的实施例)这4种方法进行比较。作为评价指标,使用实测值和预测值的r2(相关系数的平方),利用二折交叉验证法进行评价。
图7是表示在本发明的一个实施例中,样本量充分地大的情况的5的质的性状的精度评价结果的图。对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)、(4)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=10、qraf=1的情况(有遗传结构分解;本发明的实施例)这4种方法进行比较。作为评价指标,使用auc,利用二折交叉验证法进行评价。
具体实施方式
本发明的目的、特征、优点及其构思通过本说明书的记载,对本领域技术人员而言是清楚的,根据本说明书的记载,只要是本领域技术人员,就能够容易地再现本发明。以下所记载的发明的实施方式和具体的实施例等示出本发明的优选的实施方式,是为了例示或说明而示出的,但本发明并不限定于这些。在本说明书中所公开的本发明的意图以及范围内,基于本说明书的记载,能够各种各样地进行变更,这对本领域技术人员而言是清楚的。
本发明的性状预测模型制作方法为使用在生物的多个个体中使多个单核苷酸多态性(singlenucleotidepolymorphism;snp)与形状对应的单核苷酸多态性数据制作预测多因子性性状的表现型的预测模型的预测模型制作方法,其包括:将多个单核苷酸多态性分别矩阵表示的工序;将多个单核苷酸多态性基于遗传结构分类为多个类别的工序;对于各个类别,使用单核苷酸多态性的矩阵表示和属于各类别的单核苷酸多态性的个数计算基因组相似度矩阵的工序;和将基因组相似度矩阵和遗传结构的参数应用于线性混合模型的工序。或者,为一种性状预测模型制作方法,使用在生物的多个个体中使多个单核苷酸多态性、性别、年龄与形状对应的单核苷酸多态性数据,制作预测多因子性性状的表现型的性状预测模型,该包括:将多个单核苷酸多态性分别矩阵表示的工序;将性别和/或年龄矩阵表示的工序;使用单核苷酸多态性的矩阵表示和单核苷酸多态性的个数计算基因组相似度矩阵的工序;和将基因组相似度矩阵、和性别和/或年龄的矩阵应用于线性混合模型的工序。
这里使用的单核苷酸多态性数据中所含的单核苷酸多态性没有特别限定,可以是作为对象的性状的感受性多态性,也可以不是。使用的单核苷酸多态性的数量、种类也没有特别限定,优选网罗在作为对象的生物个体集合中存在1%以上的单核苷酸多态性。
作为对象的生物没有特别限定,可以为植物,也可以为动物,优选为脊椎动物,更优选为哺乳类,最优选为人。作为对象的性状只要是多因子性性状,就没有特别限定,例如,在人的情况下,能够例示与身高、体重、bmi等体型有关的指标;血压(最高血压、最低血压)、hbalc、红细胞数、血色素、血细胞容积、白细胞数、血小板数、嗜中性粒细胞的比例、淋巴细胞的比例、单核细胞的比例、嗜酸性粒细胞的比例、嗜碱性粒细胞的比例、大型不染色细胞的比例、有核红细胞数、ast(got)、alt(gpt)、γ-gtp、总胆固醇、中性脂肪、hdl胆固醇、ldl胆固醇、肌酐、尿素氮、估计肾小球过滤量、尿酸等血液检查评价值;记忆力、理解力、智商指数、运动技术等能力;肥胖·糖尿病·高血压·循环器官疾病等成人病、癌、过敏症·自免疫病等免疫疾病等病的患病容易程度等。
使用本发明的预测模型制作方法,能够进行从多个单核苷酸多态性数据预测生物个体的性状的性状预测。即,使用学习用数据集,按照本发明的性状预测模型制作方法制作性状预测模型,确定线性混合模型的参数和潜在变量,将生物个体的多个单核苷酸多态性数据应用于性状预测模型,由此,能够预测该生物个体的性状。
下面,对本发明的预测模型制作方法和性状预测方法,一边列举实施例,一边具体且详细地进行说明,但本发明并不限定于这些实施方式或实施例。
(1)性别、年龄信息的矩阵表示
阐述对n个人设为可得到性别年龄数据、作为n×6矩阵x表示的方法。x的行向量是指各个人的性别和年龄的信息。将矩阵x的i行j列要素记为x(i,j)。年龄作为类别数据使用,该类别的段数没有特别限定。这里,作为一例,对设为39岁以下、40岁以上49岁以下、50岁以上59岁以下、60岁以上69岁以下、70岁以上的5段的方法进行阐述。
性别的信息在矩阵x的第1列表示。在第i个人的个人性别为男性的情况下记为“m”、为女性的情况下记为“f”时,x(i,1)用以下的式子定义。
年龄的信息以矩阵x的第2~6列表示。将第i个人的个人年龄记为agei时,x(i,2)、x(i,3)、x(i,4)、x(i,5)、x(i,6)用以下的式子定义。
(2)基因组信息的矩阵表示
阐述对n个人设为可得到p个单核苷酸多态性(snp)数据、作为n×p矩阵(n、p为1以上的整数)w表示的方法。w的行向量是指各个人的多态性信息(profile),w的列向量是指表示某个多态性部位的个人间的不同的向量。
第i个的个人的第j个多态性由2个等位基因构成。在两个等位基因与人代表序列一致的情况下记为“aa”,在仅一个等位基因与人代表序列一致的情况下记为“ab”,在两个等位基因与人代表序列不一致的情况下记为“bb”。另外,将矩阵w的i行j列要素记为w(i,j)。另外,将第j个多态性的等位基因频率记为fj。基于这些标记,w(i,j)用以下的式子定义。
这里,关于代表序列,对各多态性确定任意的碱基,设为具有这些的序列,例如,可以为作为基因组工程的成果发表的序列。
(3)基于遗传结构的snp的分类
以下,阐述将p个snp基于遗传结构分类为多个类别的方法。表示遗传结构的具体的参数包括作为表示与性状的关联性的强度的参数的效应量(effectsize)、和表示snp的人的集合中的频率的等位基因频率(allelefrequency)。作为效应量的代表的具体例,可以列举相对危险度(relativerisk)、比值比(oddsratio)、方差贡献率、回归系数(regressioncoefficient)。等位基因频率中能够例示危险等位基因频率(riskallelefrequency;raf)或最小等位基因频率(minorallelefrequency;maf)。本发明的方法中使用的遗传结构参数没有特别限定,这里,作为一例,示出使用回归系数和raf的情况的分类步骤。
(4)分解步骤(1)效应量的qes分位数的计算
对正的整数qes,计算将分布进行了qes等分的(qes-1)个的值。以下,表示具体的分位数的计算方法,但是分位数的计算方法不限定于此。将对snp的效应量以升序排列好的数据记为es1≤es2≤…≤esp时,第i个qes分位数
这里,
(5)分解步骤(2)raf的qraf分位数的计算
对正的整数qraf,计算将分布进行了qraf等分的(qraf-1)个的值。以下,表示具体的分位数的计算方法,但是分位数的计算方法不限定于此。将对snp的raf以升序排列好的数据记为raf1≤raf2≤…≤rafp时,第j个qraf分位数
这里,
(6)snp的分类
使用通过上述步骤计算得到的
catk=(ik,jk)
(7)遗传结构参数的推定
效应量、raf等遗传结构参数能够通过多态性和性状的关联分析来推定。多态性和性状的关联分析使用通常能够获得的程序进行即可,例如,可以使用在因特网上能够获得的plink或gcta。
(8)基因组相似度矩阵的计算
基因组相似度矩阵为表示基于基因组信息的个人间的相似度的n×n矩阵。基因组相似度矩阵设为对每个qes×qraf的类别进行计算的基因组相似度矩阵。以下,示出代表性的基因组相似度矩阵a的计算式,但基因组相似度矩阵的计算式并不限定于此。
这里,a(i,j)是指类别(i,j)的基因组相似度矩阵(n×n维(次元)),p(i,j)是指属于类别(i,j)的snp的个数,w(i,j)是指从矩阵w仅切出属于类别(i,j)的snp的列向量的部分矩阵(n×p(i,j)维),w(i,j)′是指矩阵w(i,j)的转置矩阵。
(9)向线性混合模型的应用
(9-1)使用遗传结构的情况
作为使用基因组信息的预测模型,用以下的式子表示线性混合模型。
y=μ1n+g+ε
这里,y是指性状向量(n维),μ是指性状的平均值,ln是指由l构成的列向量(n维),g是指遗传因素对性状的贡献向量(n维),ε是指剩余向量(n维),g(i,j)是指属于类别(i,j)的snp向性状的贡献向量(n维),a(i,j)是指与类别(i,j)对应的基因组相似度(n×n维),i是指单元矩阵(n×n维),
(9-2)使用性别年龄信息的情况
作为使用性别年龄信息的预测模型,用以下的式子表示线性混合模型。
y=μ1n+xβ+g+ε
这里,y是指性状向量(n维),μ是指性状的平均值,ln是指由l构成的列向量(n维),x是指包含性别年龄信息的矩阵(n×6维),β是指对性别或年龄变量的重要性(6维),g是指遗传因素对性状的贡献向量(n维),ε是指剩余向量(n维),g(i,j)是指属于类别(i,j)的snp向性状的贡献向量(n维),a是指qes=1、qraf=1时的基因组相似度(n×n维),i是指单元矩阵(n×n维),
(9-3)使用遗传结构和性别年龄信息的情况
作为使用基因组信息和性别年龄信息的预测模型,用以下的式子表示线性混合模型。
y=μ1n+xβ+g+ε
这里,y是指性状向量(n维),μ是指性状的平均值,ln是指由l构成的列向量(n维),x是指包含性别年龄信息的矩阵(n×6维),β是指对性别或年龄变量的重要性(6维),g是指遗传因素对性状的贡献向量(n维),ε是指剩余向量(n维),g(i,j)是指属于类别(i,j)的snp向性状的贡献向量(n维),a(i,j)是指与类别(i,j)对应的基因组相似度(n×n维),i是指单元矩阵(n×n维),
(10)线性混合模型的参数推定
线性混合模型的参数(μ、β、
以下,将推得的参数记为
(11)贡献率的推定
使用reml的参数推定值
另外,利用以下的式子定义全部snp的贡献率的总和vg/vp。
(12)遗传因素的贡献的预测
线性混合模型的参数潜在变量(g、g(i,j)、ε)不包含在reml似然函数中,无法推定,但是能够通过以下的式子来预测。
这里,p是指通过
以下,将预测得到的潜在变量记为
(13)性状预测方法
使用上述性状预测模型,
从具备全部基因组信息、性别年龄信息、性状信息的名为nt的学习用数据集(yt、xt、wt),通过上述方法得到参数的推定值
这里,wt(i,j)是指从学习用数据集的基因组信息矩阵wt切出的仅属于类别(i,j)的snp列向量的部分矩阵(nt×p(i,j)维),a(i,j)是指从wt(i.j)计算的基因组相似度矩阵(nt×nt维),
作为式(1)的特殊例,可以考虑以下的式子(2)、(3)。
式(2)是仅使用性别年龄信息的性状预测式,式(3)是仅使用基因组信息的性状预测式。另外,在qes=1、qraf=1时,作为式(1)、式(3)的特殊例,可以分别考虑以下的式子(4)、(5)。
将式(1)称为“遗传结构分解+性别年龄调整法”,将式(2)称为“性别年龄调整法”,将式(3)称为“遗传结构分解法”,将式(4)称为“遗传结构非分解+性别年龄调整法”,将式(5)称为“遗传结构非分解法”。
(14)性状预测系统
为了将上述性状预测方法进行自动化,能够程序化,使得能够在计算机执行。这样制作得到的程序也在本发明的权利范围内。
进而,也能够形成具备用于执行该程序的计算机以及用于输入单核苷酸多态性、性别·年龄信息等的输入装置和用于输出通过程序的执行而得到的结果的输出装置的性状预测系统。
实施例
以下记载的本实施例的单核苷酸多态性信息利用humanomniexpressexome芯片(illumina公司)进行测定。
实施例1
(方法)
本实施例中,作为多因子性的量的性状的一例,着眼于身高,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据及性别·年龄信息,通过本发明的性状预测模型制作方法制作性状预测模型(使用上述(9-2)性别年龄信息的情况),推定遗传率。作为对照,对未使用性别、年龄信息的情况,也计算遗传率的推定值,与使用性别、年龄信息的情况进行比较。
接着,分别对(1)仅使用性别·年龄信息的情况、(2)仅使用单核苷酸多态性信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息两者的情况(本发明的实施例),利用二折交叉验证法评价性状预测模型的预测精度。作为评价指标,使用实测值和预测值的r2(相关系数的平方)。
(遗传率的推定方法)
在qes=1、qraf=1的情况下,将性状的方差中可以由遗传因素说明的方差的比例称为遗传率h2。关于遗传率
(结果)
未使用性别·年龄信息的情况下的遗传率为40.67%,使用性别、年龄信息的情况下的遗传率为82.29%,可知,与未使用性别·年龄信息的情况相比,在使用性别·年龄信息的情况下,遗传率大大提高,身高的方差的一部分可以由性别年龄说明。
关于(1)~(3)的3种情况,利用二折交叉验证法评价预测精度(r2)(平均±标准偏差),结果为(1)56.89±1.36%、(2)1.45±0.26%、(3)59.63±1.24%,与仅使用性别年龄信息的情况、仅使用基因组信息的情况相比,使用性别年龄信息及基因组信息这两者的情况下,预测精度提高。
实施例2
(方法)
本实施例中,作为多因子性的质的性状的一例,着眼于糖尿病的患病,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据及性别·年龄信息,通过本发明的性状预测模型制作方法制作性状预测模型(使用上述(9-2)性别年龄信息的情况)。这里,根据hbalc检查值,在其为6.5以上的情况下判定为患有糖尿病,在其低于6.5的情况下判定为没患有糖尿病。分别对(1)仅使用性别·年龄信息的情况、(2)仅使用单核苷酸多态性信息的情况、(3)使用单核苷酸多态性信息和性别、年龄信息两者的情况(本发明的实施例),利用二折交叉验证法评价性状预测模型的预测精度。作为评价指标,使用auc。
(结果)
为(1)61.39±1.56%、(2)55.76±0.28%、(3)62.98±0.61%,与仅使用性别年龄信息的情况、仅使用基因组信息的情况相比,使用性别年龄信息及基因组信息这两者的情况下,预测精度提高。
实施例3
(方法)
本实施例中,作为多因子性的量的性状的一例,着眼于hbalc检查值和身高,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据,利用遗传结构分解法进行贡献率的推定。实施(1)qes=50、qraf=1的情况、(2)qes=1、qraf=30的情况这2种情况。
(结果)
在图1中表示(1)qes=50、qraf=1的情况下的贡献率的推定结果。推定在hbalc检查值和身高的任一个中,显示中程度的效应量的单核苷酸多态性的贡献率大,显示小的效应量的单核苷酸多态性的贡献率非常小。另外,推定在hbalc检查值中,显示大的效应量的单核苷酸多态性的贡献大,但在身高中显示大的效应量的单核苷酸多态性的贡献为有限的。
在图2中表示(2)qes=1、qraf=30的情况的贡献率的推定结果。推定在hbalc检查值中,不稀有的单核苷酸多态性的贡献率为有限的,稀有的单核苷酸多态性显示非常大的贡献率。另一方面,推定在身高中,稀有的单核苷酸多态性的贡献率不小,但是不稀有的单核苷酸多态性的贡献率也不小。
实施例4
(方法)
在以充分的样本量进行学习的情况下,显示通过遗传结构分解法实现性状预测精度的提高,因此,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据及hbalc检查值,用验证用数据集进行效应量、等位基因频率的推定和线性混合模型的参数推定,用学习用数据集进行遗传因素的贡献的预测和对单核苷酸多态性的重要性的计算,用验证用数据集进行预测精度的验证。由此,能够评价假定样本量充分大的情况下的预测精度。
分别对(1)qes=1、qraf=1的情况(没有遗传结构分解)、(2)qes=10、qraf=1的情况(有遗传结构分解;本发明的实施例),利用二折交叉验证法评价性状预测模型的预测精度。作为评价指标,使用实测值和预测值的r2(相关系数的平方)。
(结果)
为(1)4.52±0.16%、(2)16.52±0.30%,假定充分的样本量的情况下,与没有遗传结构分解相比,如果有遗传结构分解,则显示预测精度显著地提高。
实施例5
(方法)
本实施例中,以图3所示的27个量的性状和5个质的性状为对象,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据,通过本发明的性状预测模型制作方法制作性状预测模型(使用上述(9-3)遗传结构及性别年龄信息的情况)。分别对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例),利用二折交叉验证法评价性状预测模型的预测精度。作为评价指标,在以量的数据为对象的情况下,使用实测值和预测值的r2(相关系数的平方),在以质的数据为对象的情况下,使用auc。
(结果)
在图4表示对27个量的性状进行了精度评价的结果,在图5表示对5个质的性状进行了精度评价的结果。关于图4、图5所示的全部27个量的性状及5个质的性状,显示与(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况相比,(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)的预测精度提高。
实施例6
(方法)
在以充分的样本量进行学习的情况下,显示通过使用性别、年龄信息或单核苷酸多态性信息和性别、年龄信息这两者而实现性状预测精度的提高,因此,以图3所示的27个量的性状及5个质的性状为对象,使用由tohokumedicalmegabankproject在平成25年所收集的4992名的单核苷酸多态性数据,通过本发明的性状预测模型制作方法制作性状预测模型(使用上述(9-3)遗传结构及性别年龄信息的情况)。分别对(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况、(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)、(4)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=10、qraf=1的情况(有遗传结构分解;本发明的实施例),利用二折交叉验证法评价性状预测模型的预测精度。作为评价指标,在以量的数据为对象的情况下,使用实测值和预测值的r2(相关系数的平方),在以质的数据为对象的情况下,使用auc。用验证用数据集进行效应量、等位基因频率的推定及线性混合模型的参数推定,用学习用数据集进行遗传因素的贡献的预测及对单核苷酸多态性的重要性的计算,用验证用数据集进行预测精度的验证。
(结果)
在图6中表示对27的量的性状进行了精度评价的结果,在图7中表示对5的质的性状进行了精度评价的结果。关于图6、图7所示的全部27个量的性状及5个质的性状,显示:与(1)仅使用单核苷酸多态性信息、且qes=1、qraf=1的情况(没有遗传结构分解)、(2)仅使用性别·年龄信息的情况相比,(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)的预测精度提高。在将(3)使用单核苷酸多态性信息和性别·年龄信息这两者、且使用qes=1、qraf=1的情况(没有遗传结构分解;本发明的实施例)与(4)使用单核苷酸多态性信息和性别·年龄信息这两者、且qes=10、qraf=1的情况(有遗传结构分解;本发明的实施例)进行比较的情况下,在全部的性状中,(4)的预测精度提高。
(结论)
如上所述,如果使用通过本发明的性状预测模型制作方法制作的性状预测模型,与现有的预测方法相比,能够以高的准确率预测性状。此外,通过利用遗传结构分解法进行贡献率的推定,能够阐明性状的遗传结构。
工业上的可利用性
根据本发明,能够提供一种用于从单核苷酸多态性数据预测性状的表现型的性状预测模型制作方法、以及能够以高的准确率预测性状的性状预测方法。
- 还没有人留言评论。精彩留言会获得点赞!