一种基于菌群优化算法的蛋白质结构从头预测方法与流程
本发明涉及一种生物学信息学、智能优化、计算机应用领域,尤其涉及的是,一种基于菌群优化算法的蛋白质结构从头预测方法。
背景技术:
蛋白质是所有生物中最重要的大分子之一,一个细胞的干重的一半以上是蛋白质。蛋白质是由单一的肽键连接在一起的氨基酸的顺序链,这些连接的链折叠成能体现它们功能的三维结构,并调节生物体中的细胞活动,以保持生命。蛋白质分子在分子水平上具体体现了蛋白质结构和功能之间的显著关系。蛋白质在生物学中有许多不同的功能。蛋白质的三维天然结构决定它的生物学机制,而反过来,蛋白质氨基酸单体的一维链又对它的天然结构进行编码。蛋白质的三维结构在药物设计、蛋白质工程和生物技术中具有决定性的重要,因此,蛋白质结构预测是一个重要的研究问题。
蛋白质结构实验测定方法包括x射线晶体学、核磁共振光谱、和电子显微镜等,这些方法被广泛用于蛋白质结构测定。x射线晶体学被认为是这些方法中相对可行且准确的测定方法之一。然而,x射线晶体学需要进行一个复杂的结晶过程,而对于一些不易结晶的蛋白(如膜蛋白),此方法无法用于结构测定。此外,这些实验测定方法极其耗时,代价昂贵,而且容易发生错误。
anfinsen热力学假说表明:蛋白质的空间结构由氨基酸序列唯一确定;蛋白质的空间结构是稳定的;蛋白质的天然构象处于自由能最低点。根据anfinsen原则,以计算机为工具,运用适当的算法,从氨基酸序列出发直接预测蛋白质的三维结构,成为当前生物信息学中一种主要的研究课题。在上世纪文献中,通常将预测方法划分为以下三类:针对高相似序列的同源建模方法;针对较低相似性序列的折叠识别方法;以及不依赖模板的从头预测方法。从头预测方法直接基于anfinsen假说建立蛋白质物理或知识能量模型,然后设计适当优化算法求解最小能量构象。一方面,从生物学意义上来讲,有助于揭示蛋白质折叠机理,进而能够最终阐明生物学中心法则中的第二遗传密码理论部分;另一方面,从现实意义来讲,该方法具有普遍性,对于序列相似度<20%或寡肽(<10个残基的小蛋白)来说,从头预测方法是唯一的选择。
目前主要的技术瓶颈在于两个方面,第一方面在于采样方法,现有技术对构象空间采样能力不强,另一方面在于构象更新方法,现有技术对构象的更新精度仍然不足。因此,现有的构象空间搜索方法存在不足,需要改进。
技术实现要素:
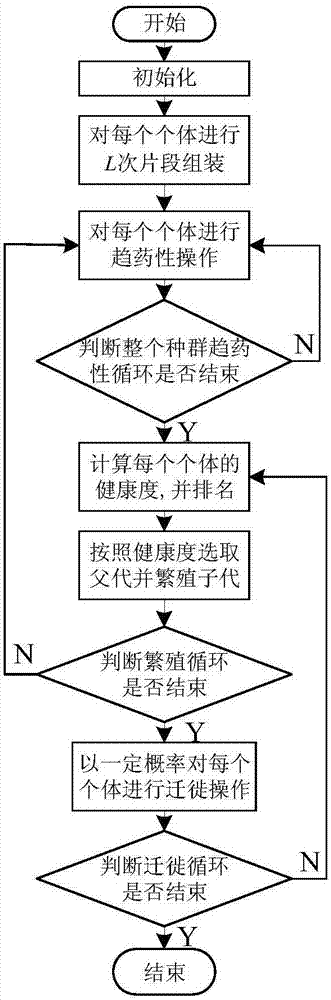
为了克服现有的蛋白质结构预测方法在构象空间搜索方面的不足,本发明提出一种基于菌群优化算法的蛋白质结构从头预测方法。根据菌群具有趋药性和聚集的特性,以及菌群优胜劣汰的自然规律,构象二面角在随机方向上前进并接受能量低的构象;待构象聚集后,淘汰一半能量高的构象,能量低的构象繁殖生成子代。此外,为了尽量避免个体陷入局部极小和过早收敛,采用迁移机制对个体进行片段组装,保证群体的多样性,有效提高构象空间采样能力,减少计算代价。
本发明解决其技术问题所采用的技术方案是:
一种基于菌群优化算法的蛋白质结构从头预测方法,所述方法包括以下步骤:
1)选取rosettascore3作为蛋白质力场模型,即能量函数j(c),c表示蛋白质构象;
2)给定输入序列信息,通过免费网络服务器robettaserver获取目标蛋白片段库;
3)参数初始化:设置细菌的种群规模np,np为偶数,第i个细菌进行趋药性行为的步长stepi,i∈{1,...,np},细菌趋药性行为循环次数nc,趋药性循环中最大前进次数ns,细菌进行繁殖行为次数nre,细菌进行迁徙行为的次数ned,迁徙概率ped;
4)用二面角对
5)构象初始化:根据给定输入序列,生成np个伸展链构象,作为初始细菌群体p={c1,c2,...,cnp},依次对细菌群体中的构象ci进行l次随机片段组装,片段从目标蛋白的片段库中随机选择;
6)趋药性行为,过程如下:
6.1)计算个体ci的能量值,并记录历史最优能量值jlast=j(ci);
6.2)随机产生维数为2l的方向向量βi,决定个体的前进方向;
6.3)根据
6.4)聚集行为,过程如下:
6.4.1)根据
6.4.2)根据
6.4.3)整个细菌种群对第i个个体的聚集作用即吸引力和排斥力之和
6.4.4)修正j(c),
6.5)令新构象
6.6)对下一个细菌进行趋药性操作6.1)~6.5),直至整个种群完成一次趋药性过程;
6.7)判断趋药性迭代过程是否结束,如果j<nc,返回步骤6),继续对细菌种群进行趋药性迭代过程,j表示当前趋药性迭代次数;否则执行下一步;
7)繁殖循环,过程如下:
71)根据
7.2)取前np/2个个体作为父代,健康度不高的后np/2个个体死亡;生成np/2个与父代完全相同的子代;
7.3)父代和子代重新编号,若k<nre,对整个种群进行趋药性操作,返回步骤6),否则,执行下一步;
8)为避免陷入局部最小和早熟,执行迁徙过程:对每个细菌个体,随机产生[0,1]的随机数rand,若rand<ped,则对该构象进行2l次随机片段组装,保证种群的多样性;
9)若l<ned,对种群执行繁殖操作,返回步骤7),否则,输出结果并退出。
本发明的技术构思为:根据菌群具有趋药性和聚集的特性,以及菌群优胜劣汰的自然规律,对构象二面角在随机方向上前进,并接受能量低的构象;待构象聚集后,淘汰一半能量高的构象,利用能量低的构象繁殖生成子代;此外,为了尽量避免个体陷入局部极小和过早收敛,采用迁移机制对个体进行片段组装,保证群体的多样性,从而有效提高构象空间采样能力。
本发明的有益效果为:本发明在蛋白质结构预测中应用,可以有效提高构象空间的采样能力,减少计算代价。
附图说明
图1是基于菌群优化算法的蛋白质结构从头预测方法对目标蛋白质1acf预测结构的三维示意图。
图2是蛋白质结构的示意图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1,一种基于菌群优化算法的蛋白质结构从头预测方法,包括以下步骤:
1)选取rosettascore3作为蛋白质力场模型,即能量函数j(c),c表示蛋白质构象;
2)给定输入序列信息,通过免费网络服务器robettaserver获取目标蛋白片段库;
3)参数初始化:细菌的种群规模np,np为偶数,第i个细菌进行趋药性行为的步长stepi,i∈{1,...,np},细菌趋药性行为循环次数nc,趋药性循环中最大前进次数ns,细菌进行繁殖行为次数nre,细菌进行迁徙行为的次数ned,迁徙概率ped;
4)用二面角对
5)构象初始化:根据给定输入序列,生成np个伸展链构象,作为初始细菌群体p={c1,c2,...,cnp},依次对细菌群体中的构象ci进行l次随机片段组装,片段从目标蛋白的片段库中随机选择;
6)趋药性行为,过程如下:
6.1)计算个体ci的能量值,并记录历史最优能量值jlast=j(ci);
6.2)随机产生维数为2l的方向向量βi,决定个体的前进方向;
6.3)根据
6.4)聚集行为,过程如下:
6.4.1)根据
6.4.2)根据
6.4.3)整个细菌种群对第i个个体的聚集作用即吸引力和排斥力之和,
6.4.4)修正j(c),
6.5)令新构象
6.6)对下一个细菌进行趋药性操作6.1)~6.5),直至整个种群完成一次趋药性过程;
6.7)判断趋药性迭代过程是否结束,如果j<nc,返回步骤6),继续对细菌种群进行趋药性迭代过程,j表示当前趋药性迭代次数;否则执行下一步;
7)繁殖循环,过程如下:
7.1)根据
7.2)取前np/2个个体作为父代,健康度不高的后np/2个个体死亡;生成np/2个与父代完全相同的子代;
7.3)父代和子代重新编号,若k<nre,对整个种群进行趋药性操作,返回步骤6);否则,执行下一步;
8)为尽量避免陷入局部最小和早熟,执行迁徙过程:对每个细菌个体,随机产生[0,1]的随机数rand,若rand<ped,则对该构象进行2l次随机片段组装,保证种群的多样性;
9)若l<ned,对种群执行繁殖操作,返回步骤7);否则,输出结果并退出。
本实施例以目标蛋白质1acf为实施例,一种基于菌群优化算法的蛋白质结构从头预测方法,所述方法包括以下步骤:
1)选取rosettascore3作为蛋白质力场模型,即能量函数j(c),c表示蛋白质构象;
2)给定输入序列信息,通过免费网络服务器robettaserver获取目标蛋白片段库;
3)参数初始化:细菌的种群规模np=50,第i个细菌进行趋药性行为的步长stepi=30,i∈{1,...,np},细菌趋药性行为循环次数nc=50,趋药性循环中最大前进次数ns=50,细菌进行繁殖行为次数nre=5,细菌进行迁徙行为的次数ned=2,迁徙概率ped=0.2;
4)用二面角对
5)构象初始化:根据给定输入序列,生成np个伸展链构象,作为初始细菌群体p={c1,c2,...,cnp},依次对细菌群体中的构象ci进行l次随机片段组装;
6)趋药性行为,过程如下:
6.1)计算个体ci的能量值,并记录历史最优能量值jlast=j(ci);
6.2)随机产生维数为2l的方向向量βi,决定个体的前进方向;
6.3)根据
6.4)聚集行为,过程如下:
6.4.1)根据
6.4.2)根据
6.4.3)整个细菌种群对第i个个体的聚集作用即吸引力和排斥力之和,
6.4.4)修正j(c),
6.5)令新构象
6.6)对下一个细菌进行趋药性操作6.1)~6.5),直至整个种群完成一次趋药性过程;
6.7)判断趋药性迭代过程是否结束,如果j<nc,返回步骤6),继续对细菌种群进行趋药性迭代过程,j表示当前趋药性迭代次数;否则执行下一步;
7)繁殖循环,过程如下:
7.1)根据
7.2)取前np/2个个体作为父代,健康度不高的后np/2个个体死亡;生成np/2个与父代完全相同的子代;
7.3)父代和子代重新编号,若k<nre,对整个种群进行趋药性操作,返回步骤6);否则,执行下一步;
8)为尽量避免陷入局部最小和早熟,执行迁徙过程:对每个细菌个体,随机产生[0,1]的随机数rand,若rand<ped,则对该构象进行2l次随机片段组装,保证种群的多样性;
9)若l<ned,对种群执行繁殖操作,返回步骤7);否则,输出结果并退出。
以上阐述的是本发明给出的一个实施例表现出来的优良效果,显然本发明不仅适合上述实施例,在不偏离本发明基本精神及不超出本发明实质内容所涉及内容的前提下可对其做种种变化加以实施。
- 还没有人留言评论。精彩留言会获得点赞!