用于变体识别的神经网络的制作方法
1.本公开涉及计算机程序和系统的领域,并且更具体地涉及关于参考基因组的变体识别(variant calling)。
背景技术:
2.在过去的几十年中,由于新基因组测序(ngs)平台所取得的技术进步,基因组测序得到了普及。随着基因组变得可用,正在开发医疗应用以对其进行利用。例如,个性化且有针对性的医疗近期越来越受到关注。直接应用包括例如遗传疾病诊断和用于治疗的癌症分析。
3.基因组测序是在硬件和软件这两个方面都在快速发展的领域,以应对精准医疗和流行病学的不断增长的需要。通常,测序经常用于确定单个个体中存在的基因组变体。基于与标准基因组参考(基因组参考联合会,genome reference consortium)的比较来标识(即“识别(call)”)变体。变体有不同的种类:在参考中替换碱基的准时变体(punctual variant)称为“snp”(单核苷酸多态性),与参考相比去除碱基的变体称为“缺失(deletion)”,并且增加碱基(即,向参考添加一个或多个碱基)的变体称为“插入(insertion)”。最后,跨越大量碱基的变体称为“结构变体”。
4.变体识别的过程可以包括检查每个基因组位置以便确定哪些等位基因存在于二倍体生发基因组中:(i)如果两个等位基因相同,则为纯合的,或(ii)如果等位基因不同,则为杂合的。每个等位基因可以与参考基因组匹配,否则是一个变体。对于肿瘤的情况,如果基因组位置与参考或位于同一位置的种系等位基因中的任一个不匹配,则变体被识别。
5.对天然dna直接进行片段化和测序或在预扩增步骤之后对天然dna进行片段化和测序,以产生读数。读数长度从100bp(例如,使用illumina技术)到10kbp(例如,使用nanopore技术)变化。这些技术的共同特性中的一个是使用冗余测序来抵消实验错误并解释细胞群中的遗传异质性。每个位置的读数的平均数量称为测序的“深度”。
6.变体识别是一个活跃的研究领域,因为具有准确且高性能的算法来开发人口规模的基因组学至关重要。已经改进了性能并且在不久的将来可能会继续改进。特别是近期,生物信息学研究人员已经试图利用机器学习方法。google开发的deepvariant尤其是这种情况。需要注意的一点是,这个问题对于正常dna来说是相当困难的,但对于肿瘤dna来说更具挑战性。事实上,肿瘤细胞突变非常迅速,因此它们不共享相同的dna:它是一个异质的群体。
7.在该上下文内,仍然需要一种用于关于参考基因组进行变体识别的改进的解决方案。
技术实现要素:
8.因此,提供了一种用于对神经网络进行机器学习以关于参考基因组进行变体识别的计算机实现的方法。神经网络被配置为取一组或多组数据片段作为输入,每组数据片段
指定相对于参考基因组的基因组位置对齐的相应的读数。神经网络被配置为输出关于在基因组位置处变体的存在的信息。对于每组数据片段,神经网络包括相应的函数,该相应的函数被配置为取该组数据片段作为输入并且处理该组数据片段。该相应的函数是对称的。
9.机器学习方法可以包括以下各项中的一项或多项:
[0010]-每个数据片段包括相应的读数的相应的碱基描述符的序列;
[0011]-每个相应的函数包括一个或多个卷积层,该一个或多个卷积层用于处理每个相应的碱基描述符的序列;
[0012]-每个卷积层应用一个或多个一维卷积滤波器;
[0013]-碱基描述符包括表示插入大小和/或删除大小的一个或多个描述符,例如,插入大小描述符和删除大小描述符;
[0014]-每个相应的函数包括归约层,该归约层用于将一个或多个卷积层的输出与次序无关地折叠为一组特征;
[0015]-归约层进一步取针对每个相应的读数的读数描述符作为输入;
[0016]-读数描述符包括单倍型(haplotype)支持描述符;
[0017]-归约层包括一个或多个与次序无关的算子,例如,均值和/或标准差;
[0018]-神经网络包括一个或多个全连接层,该一个或多个全连接层被配置为处理每个归约层的输出并且执行分类;
[0019]-神经网络进一步取堆(pile)描述符作为输入;
[0020]-堆描述符包括表示深度的描述符和/或表示贝叶斯变体估计的描述符;和/或
[0021]-一组或多组读数包括用于种系变体的第一组读数和用于体细胞变体的第二组读数,神经网络包括:
[0022]
ο用于第一组读数的第一函数和用于第二组读数的第二函数,以及
[0023]
ο用于聚合第一函数和第二函数的输出的层。
[0024]
进一步提供了一种用于关于参考基因组进行变体识别的第一计算机实现的方法。第一变体识别方法包括提供一组或多组数据片段作为输入,每组数据片段指定相对于参考基因组的基因组位置对齐的相应的读数。第一变体识别方法包括将根据机器学习方法进行机器学习的神经网络应用于输入,以便输出关于在基因组位置处变体的存在的信息。神经网络的应用包括对于每组数据片段,应用神经网络的相应的对称函数,该相应的对称函数被配置为取该组数据片段作为输入并且处理该组数据片段。
[0025]
进一步提供了一种用于关于参考基因组进行变体识别的第二计算机实现的方法,其包括第一变体识别方法。第二变体识别方法包括提供相对于参考基因组对齐的一组或多组读数。第二变体识别方法包括通过将一组或多组读数与参考基因组进行比较来确定参考基因组中的一组感兴趣区域。对于每个给定的感兴趣区域,第二变体识别方法包括以下步骤。第一步骤是基于给定区域的所提供的一组或多组读数执行单倍型重建,以确定两个或更多个单倍型。第二步骤是基于两个或更多个单倍型重新对齐给定区域的一组或多组读数。第三步骤是基于重新对齐的一组或多组读数和两个或更多个单倍型来推断给定区域的潜在变体。第四步骤是执行粗粒度过滤以从潜在变体中检测候选变体。每个检测到的候选变体对应于相应的基因组位置。在第五步骤中,对于每个检测到的候选,第二变体识别方法包括以下子步骤。第一子步骤是确定一组或多组数据片段,每组数据片段指定相对于基因
组位置对齐的相应的读数,该基因组位置对应于检测到的候选。第二子步骤是利用一组或多组数据片段来执行第一变体识别方法的提供和应用。
[0026]
第二变体识别方法可以包括以下各项中的一项或多项:
[0027]-单倍型重建的执行包括:
[0028]
ο通过在有向无环图中枚举预定数量的最长路径来推理一组潜在单倍型;并且可选地
[0029]
ο从该组潜在单倍型中选择单倍型的子集,该单倍型的子集是该组中具有最高数量的支持读数的潜在单倍型,该单倍型的子集对应于单倍型重建确定的两个或更多个单倍型;
[0030]-一组或多组读数包括用于种系变体的第一组读数,对给定区域的潜在变体的推断包括:对于种系变体,评估变体比参考更有可能的概率;
[0031]-一组或多组读数包括用于体细胞变体的第二组读数,对给定区域的潜在变体的推断考虑对于体细胞变体的以下各项:
[0032]
ο种系变体的存在和/或体细胞变体的存在,以及
[0033]
ο体细胞变体频率;
[0034]
进一步提供了一种数据结构,包括根据机器学习方法(即,表示神经网络的数据)进行机器学习的神经网络。
[0035]
进一步提供了一种计算机程序,包括用于执行机器学习方法的指令。
[0036]
进一步提供了一种计算机程序,包括用于执行第一变体识别方法和/或第二变体识别方法的指令。
[0037]
进一步提供了一种设备,包括数据存储介质,该数据存储介质上记录有数据结构和/或计算机程序中的任一个或两个。
[0038]
该设备可以形成或用作例如在saas(软件即服务)或其他服务器、或基于云的平台等上的非暂时性计算机可读介质。该设备可以可替代地包括耦合到数据存储介质的处理器。因此,该设备可以整体或部分地形成计算机系统(例如,该设备是整个系统的子系统)。该系统还可以包括耦合到处理器的图形用户接口。
附图说明
[0039]
现在将参考附图描述非限制性示例,在附图中:
[0040]-图1示出了第一变体识别方法的流程图;
[0041]-图2示出了第二变体识别方法的流程图;
[0042]-图3示出了变体识别过程的示例;
[0043]-图4示出了单倍型重建的示例;
[0044]-图5示出了作为召回(recall)的函数的种系候选变体计数的示例;
[0045]-图6示出了作为召回的函数的体细胞候选变体计数的示例;
[0046]-图7示出了堆积图像(pile-up image)的示例;
[0047]-图8示出了神经网络的架构的示例;
[0048]-图9示出了用于识别体细胞变体的神经网络的架构的示例;
[0049]-图10示出了针对种系情况的精度、召回和f1分数度量的结果的示例;
[0050]-图11示出了针对种系情况的错误数量度量的结果的示例;
[0051]-图12示出了针对体细胞情况的与现有技术变体识别器的比较结果的示例;以及
[0052]-图13示出了系统的示例。
具体实施方式
[0053]
提出了一种用于对神经网络进行机器学习以关于参考基因组进行变体识别的计算机实现的方法。神经网络被配置为取一组或多组数据片段作为输入,每组数据片段指定相对于参考基因组的基因组位置对齐的相应的读数。神经网络被配置为输出关于在基因组位置处变体的存在的信息。对于每组数据片段,神经网络包括相应的函数,该相应的函数被配置为取该组数据片段作为输入并且处理该组数据片段。该相应的函数是对称的。
[0054]
参考图1的流程图,进一步提供了一种用于关于参考基因组进行变体识别的第一计算机实现的方法。第一变体识别方法包括提供s700一组或多组数据片段作为输入,每组数据片段指定相对于参考基因组的基因组位置对齐的相应的读数。第一变体识别方法包括将根据机器学习方法进行机器学习的神经网络应用s800于输入。
[0055]
神经网络被配置为取一组或多组数据片段作为输入,并且输出关于在基因组位置处变体的存在的信息。对于每组数据片段,神经网络包括相应的函数,该相应的函数被配置为取该组数据片段作为输入并且处理该组数据片段。每组的相应的函数是对称的。神经网络的应用包括对于每组数据片段,应用神经网络的相应的对称函数,该相应的对称函数被配置为取该组数据片段作为输入并且处理该组数据片段。
[0056]
参考图2的流程图,进一步提供了一种用于关于参考基因组进行变体识别的第二计算机实现的方法,其结合了图1的方法。第二变体识别方法包括提供s000相对于参考基因组对齐的一组或多组读数。第二变体识别方法包括通过将一组或多组读数与参考基因组进行比较来确定s100参考基因组中的一组感兴趣区域。对于该组的每个给定的感兴趣区域,第二变体识别方法包括以下步骤。第一步骤在于基于给定区域的所提供的一组或多组读数执行s200单倍型重建,以确定两个或更多个单倍型。第二步骤在于基于两个或更多个单倍型重新对齐s300给定区域的一组或多组读数。第三步骤在于基于重新对齐的一组或多组读数和两个或更多个单倍型来推断s400给定区域的潜在变体。第四步骤在于执行s500粗粒度过滤以从潜在变体中检测候选变体。每个检测到的候选变体对应于相应的基因组位置。在第五步骤中,对于每个检测到的候选,第二变体识别方法包括以下子步骤。第一子步骤包括确定s600一组或多组数据片段,每组数据片段指定相对于基因组位置对齐的相应的读数,该基因组位置对应于检测到的候选。第二子步骤包括利用第一变体识别方法的一组或多组数据片段来执行提供s700和应用s800。
[0057]
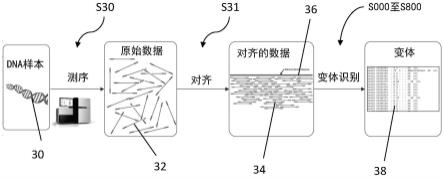
对该组区域的确定s100可以包括执行一个过程,该过程包括遍历参考基因组的位置,并且对于每个给定位置,计算在给定位置上对齐的读数中的错配率。在该步骤期间,该过程可以包括记录(即,标记)具有高于预定阈值的比率的位置。同时,该过程可能将这些位置聚合为感兴趣区域(其中错配率比阈值高几倍的那些区域)。一旦该过程计算出具有高于阈值的比率的位置,该过程就可以将该区域记录为感兴趣区域。确定s100可以包括在预定限制(例如,1000个碱基的限制)内将区域从一个碱基扩大到下一个碱基。
[0058]
这种方法形成了针对关于参考基因组进行变体识别的改进的解决方案。
[0059]
值得注意的是,机器学习方法利用了机器学习技术方法,例如,速度、准确度和/或利用可用数据。此外,机器学习方法允许高效和/或准确的机器学习。事实上,相应的函数是对称的,这允许神经网络学习侧重于基本信息,因为读数的次序在测序输出中没有任何意义。
[0060]“对称”指代对称函数的数学定义。换言之,“对称”函数是若干变量的函数,它对其变量的任何排列都是不变的(无论是σ任何排列,f(x1,
…
,xn)=f(σ(x1,
…
,xn)))。因此,输入的一个或多个组中的每一个组中的读数指定的数据片段的次序对结果没有影响,即,对关于由神经网络输出的变体的存在的信息没有影响。换言之,神经网络在构造上对于每个输入的组中的读数指定的数据片段的次序是不变的。
[0061]
在神经网络被配置为取若干组数据片段以及因此若干个函数作为输入的情况下,这些函数可以是相同的,即,具有相同的参数值和/或具有共享的加权。可替代地,函数可以是独特的/不同的。
[0062]
机器学习方法可以包括提供用于参考基因组的训练样本的数据集,以及基于所提供的训练样本的数据集来训练神经网络。每个训练样本可以包括:一组或多组数据片段,每组数据片段指定相对于参考基因组的给定的基因组位置对齐的相应的读数;以及用于神经网络的对应的输出,即,关于在给定的基因组位置处变体的存在的输出信息。这样的输出(即,地面真值(ground truth))可以经由传统的确定性技术在数据集的构建中获得。如机器学习领域本身已知的,由神经网络对输入的处理包括对输入应用操作,该操作由包括加权值的数据定义。因此,对神经网络的学习/训练包括基于被配置用于这种学习的数据集来确定加权值,这种数据集可能被称为学习数据集或训练数据集。数据集可以包括训练样本。每个训练样本可以包括相对于基因组位置对齐的相应的读数的相应的一组或多组数据片段和关于在基因组位置处变体的存在的相应的信息。相应的信息可以是一项或多项初步研究的结果,并且可以被视为用于学习的地面真值。训练样本表示其中神经网络在学习后要使用的情况的多样性。数据集可以包括高于1000、10000、100000或1000000的多个训练样本。在本公开的上下文中,“学习神经网络”意味着数据集是基于加权值(也称为“参数”)的神经网络的学习/训练数据集。
[0063]
神经网络可以由深度神经网络(dnn)组成和/或包括若干个子网络。机器学习方法可以包括联合地或单独地训练若干个子网络。“联合”意味着若干个子网络的加权都是相同的单个优化中的变量。在若干组数据片段的情况下,每个函数可以形成相应的子网络,并且机器学习方法可以包括联合地或单独地针对若干组数据片段训练所述函数或子网络。
[0064]
现在讨论神经网络的输入。
[0065]
读数是相应的dna片段的碱基序列(或等效地为碱基对序列)。本文中的任何读数可以通过基于dna测序的任何技术(例如,illumina技术、ion torrent技术或nanopore技术)对一个或多个细胞进行dna测序而获得。测序可以包括将一个或多个细胞的dna(即,天然dna)片段化为多个dna片段,并且可选地在dna扩增步骤之后基于多个dna片段来产生读数。本文中任何dna测序的一个或多个细胞可以在个体身上采样。因此,变体识别方法可以各自包括在个体身上对一个或多个细胞进行采样的初始步骤,如上对细胞进行dna测序以便产生读数,然后产生要输入到神经网络的数据片段。可以以任何方式和/或使用任何采样仪器执行采样以从个体采集样本。
[0066]
参考基因组可以是任何(单一)物种(特别是人类物种)的参考基因组。本文的任何读数可以是所述物种的个体的读数。换言之,读数起点处的细胞可以是所述个体的细胞。参考基因组可以由数字核酸序列数据库组成,该数字核酸序列数据库表示该物种的一个理想化个体生物体中的一组基因的示例。
[0067]
每个读数相对于基因组位置对齐,这意味着指定读数的数据片段包含读数对应于或映射到参考基因组的哪个部分的指示。
[0068]
每个数据片段形成相对于参考基因组的基因组位置对齐的相应的读数的计算机规范。换言之,每个数据片段包括描述或涉及相应的读数的信息,例如,形成读数的碱基序列,以及可选地与读数或其中的碱基相关的附加信息。例如,该信息可以包括相应的读数的碱基序列的经编码的版本。
[0069]
现在讨论神经网络的输出。
[0070]
输出的信息可以包括(即,在基因组位置处)是否存在变体的指示,以及可选地包括(即,在预定的一组变体类型中)变体的类型的指示。另外地或可替代地,输出的信息可以包括存在变体的概率。神经网络可以通过考虑每个组的所有数据片段来计算存在变体的概率。可替代地,神经网络可以仅输出变体存在的显著概率。例如,神经网络可以计算在基因组位置存在变体的概率,并且仅当该概率高于阈值时才输出该计算出的概率。输出的信息还可以包括其他信息,例如,关于基因组位置的信息、关于变体类型的信息、关于变体是纯合子还是杂合子的信息。
[0071]
一组预定的变体类型可以包括针对准时变体(即,snp)的一种或多种类型、针对缺失(即,与参考相比去除一个或多个碱基的变体)的一种或多种类型、以及针对插入(即,与参考相比增加一个或多个碱基的变体)的一种或多种类型。对于删除和插入,去除或增加的碱基的数量可能有最大数量。换言之,如果添加或去除的碱基的数量小于或等于最大数量,则神经网络可以输出变体是插入或缺失。信息可以以任何格式(例如,文本格式或表格格式)输出,例如,每个位置一行或一排被确定为具有变体或可能具有变体。
[0072]
可以训练和设计神经网络以应用于全部指定(例如,人类物种的)个体的种系读数(即,来自健康细胞的读数)的数据片段。在这种情况下,输出可以指示个体的健康细胞中是否存在变体。神经网络可以可替代地被训练和设计以应用于指定患有癌症疾病的个体的体细胞读数(即,来自癌细胞的读数)的数据片段,并且可选地与指定同一个体的种系读数的数据片段一起。在这种情况下,输出可以指示个体的体细胞中是否存在变体。后一种情况允许识别特定于某些癌症疾病的变体。
[0073]
现在讨论变体识别方法的应用。
[0074]
对于种系情况,应用可以是疾病的预后、诊断或治疗(例如,遗传疾病的诊断)。例如,该应用可以包括分析个体的识别出的变体、以及诊断疾病和/或确定用于治疗所诊断的疾病的适合的治疗(例如,剂量)和/或执行预后和/或确定适合的行为。经历变体识别方法的个体(例如,人类)然后可以遵循这样的治疗和/或行为。
[0075]
对于体细胞情况,应用可以是影响受试者个体的癌症疾病的评估或预后和治疗(例如,确定癌症的细节,或确认癌症诊断)。该应用可以包括通过比较针对健康细胞识别出的变体与针对体细胞识别出的变体来标识体细胞中的突变。该应用还可以包括基于个体的识别出的变体和所标识的突变来提供针对这种癌症的特定的治疗。可替代地或另外地,该
应用可以包括基于所标识的突变来预测癌症的演变。
[0076]
变体识别方法中的任一个可以包括显示神经网络的输出和/或上述应用的结果。
[0077]
该方法是计算机实现的。这意味着该方法的步骤(或实质上所有步骤)由至少一个计算机或任何类似系统执行。因此,该方法的步骤由计算机可能是全自动或半自动地执行。在示例中,该方法的步骤中的至少一些步骤的触发可以通过用户-计算机交互来执行。所要求的用户-计算机交互的水平可能取决于预见的自动化水平,并且与实现用户愿望的需要相平衡。在示例中,该水平可以是用户定义的和/或预定义的。
[0078]
每个数据片段可以包括相应的读数的相应的碱基描述符的序列。换言之,每个数据片段可以包括具有坐标的碱基描述符的向量,每个坐标对应于参考基因组的相应的位置,并且每个向量具有对应于对齐的基因组位置的一个坐标。碱基描述符可以包括碱基的类型(例如,a、t、c或g),并且相应的碱基描述符的序列可以包括读数的碱基序列。因此,每个数据片段的长度可以对应于读数的碱基序列的大小(即,取决于每个读数的大小是可变的)。可替代地,每个数据片段的长度可以是同质的(例如,恒定的),并且数据片段可以全部以基因组位置为中心。在这种情况下,每个数据片段还可以包括至少一个附加的起始序列或结束序列,其位于对应于读数的碱基描述符的序列的开始或结尾,并且具有变化以对齐参考基因组的上的每个读数的相应的序列的大小。每个附加的起始序列或结束序列可以由具有空值的一个或多个碱基描述符的序列组成。换言之,通过在每个向量的开始和/或结尾添加空值来对向量进行归一化,使得所有向量具有相同的长度并且所有向量具有在基因组位置上对齐的相同坐标。这种归一化有助于(例如,由卷积层)对每组碱基描述符序列进行通用处理。
[0079]
每个相应的函数可以包括一个或多个卷积层,该一个或多个卷积层用于处理每个相应的碱基描述符的序列,即,每个相应的函数将一个或多个卷积应用于每个相应的碱基描述符的序列。在神经网络被配置为取若干个组作为输入并因此包括若干个对称函数的情况下,一个或多个卷积层对于不同的函数可以是相同的,即,可以包括共享的加权。可替代地,一个或多个卷积层对于不同的函数可以是不同的。每个卷积层可以应用一个或多个一维卷积滤波器。每个一维卷积滤波器可以取表示每个数据片段的向量l
×
1作为输入并且对其进行处理,其中l是每个数据片段的长度。一维卷积滤波器的应用允许独立考虑每个读数,而不考虑读数的次序。因此提高了学习的效率和/或准确度。事实上,神经网络的学习侧重于基本信息(通过不试图从输入的读数之间的次序中学习某些东西,那里没有什么要学习的)。
[0080]
每个相应的函数可以包括归约层,该归约层用于将一个或多个卷积层的输出与次序无关地折叠为一组特征。例如,归约层可以包括一个或多个与次序无关的算子,例如,均值和/或标准差。在若干个组以及因此若干个对称函数的情况下,归约层对于不同的函数可以是相同的,即,可以包括相同的参数值(例如,相同的与次序无关的算子)。可替代地,归约层对于不同的函数可以是不同的。
[0081]
参考图3至图11以及以下第1节至第3节,现在讨论机器学习方法以及第一变体识别方法和第二变体识别方法的具体实现方式。
[0082]
第1节:变体识别过程
[0083]
图3示出了变体识别过程的示例,其中实现了变体识别方法的具体实现方式。变体
识别过程包括对(例如,人类物种,例如,患者的)一个个体的天然dna 30进行测序s30。天然dna 30可以直接被片段化和测序s30或在预扩增步骤之后被片段化和测序s30以产生读数32。对天然dna的测序可以利用dna测序的任何技术(例如,illumina技术或nanopore技术)来执行。读数长度可能取决于dna测序的技术而变化。读数长度可能从illumina技术的100bp(碱基对(base pair)的首字母缩写)到nanopore技术的10kbp(1kbp=1000bp)变化。该方法可以实现的测序技术的共同特性中的一个是使用冗余测序来抵消实验错误并解释细胞群中的遗传异质性。每个位置的读数的平均数量称为测序的深度。
[0084]
在测序s30之后,变体识别过程包括参考在基因组36上读数32的对齐s31。对齐s31可以包括例如基于已知的映射算法(例如,burrows-wheeler对齐器)的读数对参考基因组的映射。对齐s31可以包括基于碱基序列相似度将每个读数与参考基因组定位。对齐s31允许标识在每个读数的碱基序列与参考基因组的相应的碱基序列之间的相似度。由于冗余,在参考基因组的每个基因组位置上对齐了若干个读数,并且在给定的基因组位置上对齐的若干个读数形成了针对基因组位置的读数的堆。相对于参考基因组对齐的读数可以形成第一组读数。
[0085]
在变体识别过程中,对于dna的不同样本,测序s30和对齐s31步骤可以连续或并行重复。例如,测序s30和对齐s31步骤可以针对用于种系变体的第一样本和用于体细胞变体的第二样本执行。在这种情况下,该过程形成了相对于基因组位置对齐的若干组(例如,两组)读数。所形成的一组或多组读数被提供作为第二变体识别方法中的输入。
[0086]
第二变体识别方法的输出是变体列表38,该变体列表38是来自由神经网络在应用于针对每个检测到的候选变体的所提供的一组或多组读数时输出的信息的编译的结果。编译可以包括基于每个变体的概率从针对每个检测到的候选变体输出的信息中选择变体列表38。对于列表的每个变体,输出包括描述变体的信息,例如,关于变体的基因组位置的信息、关于变体的类型的信息以及关于变体是纯合子还是杂合子的信息。
[0087]
第2节:变体候选检测
[0088]
2.1单倍型重建和局部重新对齐
[0089]
单倍型重建和局部重新对齐的步骤改进了对变体的识别。实际上,单倍型的重建允许对映射到同一区域的所有读数的信息进行组合,然后使用经重建的单倍型重新对齐读数,这改进了读数与参考基因组的对齐。
[0090]
为了节省计算资源,变体识别方法将其限制在包含具有大量错配的读数的区域(即,该方法将单倍型重建限制在一组感兴趣区域)。实际上,与参考基因组存在许多差异的区域是其中可以找到变体的区域。变体识别方法在这些感兴趣区域中重建单倍型(即,执行局部染色体片段),这允许正确地重新对齐读数。
[0091]
确定一组感兴趣区域可以包括确定具有高于给定阈值的在读数与参考基因组之间的错配率的位置,并且将确定出的位置聚合为一组感兴趣区域。例如,确定具有高于给定阈值的错配率的位置可以包括连续遍历参考基因组(或参考基因组的一部分)的位置,并且对于每个给定位置,计算在这个给定的位置上对齐的读数的差异率(即,错误率)。在遍历基因组位置时,如果计算出的比率高于阈值,则该方法可以记录给定位置。依次或同时,该方法可以将记录的位置聚合为感兴趣区域。聚合可以包括选择映射到所记录的位置的相邻位置(例如,由围绕位置的每侧上的预定数量的碱基(例如,每侧100bp)定义的邻域中的位置)
的读数。
[0092]
参考图4,现在讨论单倍型重建的示例。示例的单倍型重建基于无环de-bruijn图40。示例的单倍型重建包括将每个读数拆分为具有初始k-mer大小的多个k-mer,并基于k-mer来计算初始de-bruijn图。在de-bruijn图中,节点42是k-mer,并且边44指示两个连续k-mer之间的过渡。初始k-mer大小可能在20bp到30bp之间变化,以确保最小的特异度。实际上,一方面,k-mer越小,它们的特异度就越低(因此位于不同位置的两个k-mer发生碰撞的可能性就越大),并且另一方面,小k-mer不太可能包含实验错误。因此,在20bp到30bp之间的初始k-mer大小是考虑到这两种趋势的折衷方案。然后,单倍型重建包括增加k-mer大小,直到获得的de-bruijn图是无环的。k-mer大小的增加可能是递增的,并且可以在每个新的k-mer大小处计算新的de-bruijn图。
[0093]
然后通过在计算出的无环de-bruijn图中枚举n个最长路径(例如,n=100)来推理潜在单倍型。单倍型是包括变体的碱基序列(单倍型是统计确定的)。它对应于给定患者的参考。然后,该方法从一组潜在单倍型中选择单倍型的子集。子集中的单倍型是具有最高数量的支持读数的潜在单倍型。单倍型的子集对应于单倍型重建最终确定的两个或更多单倍型。为了选择单倍型的子集,该方法通过过滤推理得出的潜在单倍型中的相关单倍型来枚举单倍型。对相关单倍型的过滤可以包括将读数与不同的单倍型重新对齐并保持读数支持的单倍型。当读数与一个单倍型比其他单倍型对齐得更好时,读数“支持”该单倍型。相关单倍型可能是具有最高数量的支持读数的单倍型。该方法可以列举两种单倍型。事实上,例如,在人类的情况下,dna可以是二倍体。可替代地,该方法还可以列举多于两个的单倍型。实际上,测序可能会提供在同一区域中的若干个变体,并且当变体相距太远时,可能由于读数和k-mer的大小有限而难以重新校正它们,这可能会导致信息丢失。通过列举两个或更多个单倍型,变体识别方法因此改进了对变体的识别。
[0094]
在列举两个或更多单倍型之后,读数可以与它们支持的单倍型重新对齐,并且单倍型可以与参考对齐。然后,通过简单的对齐组合,该方法例如基于已知算法(例如,smith-waterman-gotoh的算法)将读数与参考重新对齐。
[0095]
2.2变体贝叶斯评估(过滤)
[0096]
对于每个基因组位置,该方法可以从在单倍型重建期间建立的两个或更多单倍型的列表中推断出潜在变体。然后,该方法可以执行粗粒度过滤以从潜在变体中检测候选变体。通过使用碱基质量和读数支持信息,变体识别方法可以实现贝叶斯框架,该贝叶斯框架执行粗粒度过滤。变体识别方法可以独立地处理与一个或多个组中的每一个组相关的数据。例如,该方法可以基于全局贝叶斯计算来处理与体细胞数据相关的组(如以下节中所讨论的)。变体识别方法可以通过考虑种系变体和细胞间异质性来处理与体细胞数据相关的组。一组或多组读数包括用于种系变体的第一组读数和用于体细胞变体的第二组读数。以下节中讨论了针对每组的粗粒度过滤的执行。
[0097]
2.2.1种系
[0098]
对于给定的基因组位置,变体识别方法评估任何变体比参考更有可能的概率。任何变体比参考更有可能的概率可以基于以下公式表示:
[0099]
[0100]
其中,vari是变体位于所考虑的位置处的假设。p(vari)是考虑变体纯合性特性的vari的先验概率。p(pile|vari)是在知道给定位置处存在变体的情况下,该组特定的读数映射给定位置的概率。通过使用支持读数信息和碱基质量的单倍型来计算p(pile|vari)。p(ref|pile)是错误概率。该方法可以考虑该变体是纯合的(相同的变体存在于两对染色体上)还是杂合的(变体存在于该对的单个染色体上)。该方法可以针对每种情况计算概率。例如,纯合变体情况可以基于以下公式表示:
[0101][0102]
其中,表示支持变体i的一组读数,并且ej是根据所考虑位置的读数j的碱基质量计算出的错误概率。该方法可以基于由测序技术提供的指标来计算错误概率ej。该方法可能会忽略插入删除错误,因为与替换相比,它们的发生率要低得多。
[0103]
2.2.2体细胞
[0104]
对于体细胞情况,变体识别方法可以考虑一组假设。该方法对每个候选变体的每个基因组位置执行分析。该方法考虑了相同基因组位置上的种系变体和体细胞变体。该组包括第一假设,即,不存在种系变体和体细胞变体。该组包括第二假设,即,存在种系变体且没有体细胞变体。该组包括第三假设,即,不存在种系变体且有体细胞变体。该组包括第四假设,即,存在种系变体和体细胞变体。
[0105]
另外地,该方法可以考虑体细胞变体可能仅存在于某些细胞中。细胞中存在体细胞变体的频率是未知的,并且是该方法可能考虑的附加假设。例如,具有体细胞变体频率f的第三假设可以基于以下公式表示:
[0106]
p(pile
soma
,pile
germ
|var
soma
(f),ref
germ
)=p(pile
soma
|var
soma
(f),ref
germ
)
·
p(pile
germ
|ref
germ
)
[0107]
其中
[0108][0109]
并且
[0110][0111]
2.3性能评估
[0112]
该方法可以执行粗粒度过滤以检测候选变体。粗粒度过滤确保即使该方法保留了太多候选,也很少有真正的变体被遗漏。“召回”是一个度量,表明该方法已经丢失了多少变体。该方法可以从数据集(例如,由giab(genome in a bootle的首字母缩写)提供的数据集)中取回真正的变体。召回可以用公式召回=tp/(tp+fn)表示,其中tp表示真阳性,并且fn表示真阴性。召回的值为“0”可能意味着该方法尚未选择任何真正的变体,而召回的值为“1”可能意味着该方法尚未忘记任何变体。当召回的值很高而选择的变体候选的数量很低时,过滤会得到改进。
[0113]
图5示出了作为针对hg001和22号染色体的召回函数的种系候选变体计数的示例。该方法通过改变选择阈值(例如,在区间[-100;100]之间)来计算图。该方法可以包括基于召回的演变来选择选择阈值。虚线50对应于由本示例中的方法选择的阈值。在本示例中,对于所选择的阈值,召回等于0.997,候选的数量为44256。
[0114]
图6示出了作为针对colo-829和22号染色体的召回函数的体细胞候选变体计数的示例。对于种系情况,通过改变选择阈值获得图并且该方法可以选择选择阈值(阈值可以也在区间[-100;100]中变化)。朴素模型(naivemodel)61是种系贝叶斯模型,独立应用于种系和体细胞堆,而体细胞混合模型62是结合两个堆的贝叶斯模型。虚线63对应于由方法选择的阈值(召回等于0.934,并且候选的数量为56767)。
[0115]
2.4数据片段生成
[0116]
一旦已经确定了候选变体,变体识别方法就针对每个检测到的候选变体确定一组或多组数据片段,每组数据片段指定相对于基因组位置对齐的相应的读数,该基因组位置对应于检测到的候选变体。每个数据片段包括具有多个(例如,多于3个)独立通道的2d结构。每个数据片段还可以包括1d结构和一组标量,如以下节中所描述的。每个数据片段指定相对于基因组位置对齐的相应的读数,即,每个数据片段指定映射给定基因组位置的读数的堆中的一个读数。可以在堆积图像中示出读数的堆积。
[0117]
图7示出了堆积图像的示例。堆积图像是与给定基因组位置相交的读数(即,映射到给定基因组位置的读数)的2d表示。堆的每一行表示在基因组位置73上对齐的相应的读数。读数中的每一个与参考基因组对齐。读数中的每一个具有相应的碱基序列,当读数与参考对齐时,该碱基序列包括基因组位置73。读数可以按照基因组位置在它们相应的序列中出现的次序在堆中布置。
[0118]
图7中示出了两个不同的堆积图像。图像a是针对种系变体获得的一组读数的堆积图像,并且图像b是两组读数的堆积图像,一组是针对种系变体获得的,另一组是针对体细胞变体获得的。堆积图像表示两种信息:读数描述符73和碱基描述符74。读数描述符73位于左侧第一列。读数描述符包括体细胞读数描述符75、支持参考的读数描述符76和支持变体单倍型的读数描述符77。每个读数对于每个读数描述符75、76和77具有相应的值,其被编码在数据片段中。例如,对于体细胞读数描述符75,每个数据片段包括真值或假值,这取决于读数是针对种系还是针对体细胞变体的。
[0119]
堆积图像还表示碱基描述符74。碱基描述符74被绘制在图7中的读数表示本身上。碱基描述符74被编码在每个读数的数据片段中。对于每个读数,碱基描述符74表示在读数的相应的碱基序列与参考基因组之间的错配。碱基描述符74可以包括用于每个潜在错配的特定碱基描述符,例如,用于碱基a、t、g和c中的每一个的碱基描述符。
[0120]
现在讨论堆描述符、读数描述符和碱基描述符的示例。该方法可以实现这些描述符的全部或任何部分,并且该方法还可以可选地实现其他描述符。该方法可以例如在不同的通道上在指定每个读数的数据片段中对描述符进行编码。取决于描述符的复杂度,每个描述符可以在一个或多个独立通道上编码。
[0121]
堆描述符(分别是读数描述符或碱基描述符)各自表示堆的一个方面(分别是读数或碱基)并且由量化所述方面的值组成。在神经网络被配置为仅取单组数据片段作为输入的情况下,该组可以对应于单个读数堆,并且堆描述符可以指定所述堆。在神经网络被配置
为取若干组数据片段作为输入的情况下,一组或多组数据可以完全对应于同一读数堆,并且堆描述符可以指定所述堆。可替代地,每个组可以对应于相应的不同的读数堆或被视为相应的不同的读数堆,并且神经网络可以取指定每个这样的堆的堆描述符作为输入。堆描述符可以输入到一个或多个全连接层,例如,与每个归约层的输出聚合(例如,级联)。
[0122]
堆描述符可以包括深度描述符,其表示堆的深度(即,堆叠在堆中的读数的数量)。堆描述符可以由用于所考虑的堆的堆中的多个读数组成。该方法可以在一个通道中编码深度描述符。堆描述符还可以包括例如在四个通道(例如,取决于所考虑的假设为p00、p01、p11和p12)中编码的贝叶斯变体估计的描述符(来自预过滤的数据)。
[0123]
读数描述符可以包括体细胞描述符,其可以仅针对体细胞数据而存在。读数描述符可以包括针对单倍型的支持描述符,其描述读数支持的单倍型(例如,当读数支持参考时为h0,当读数支持第一单倍型时为h1,并且当读数支持第二单倍型时为h2)。针对单倍型的支持描述符(也称为单倍型支持描述符)可以在三个通道中编码(一个用于参考基因组,一个用于两个最受支持的单倍型中的每一个)。可选地,读数描述符可以包括映射质量描述符。映射质量由执行初始对齐的对齐器提供的数据组成。每个读数的映射质量测量相对于读数的对齐的置信水平。例如,当读数在基因组的至少两个区域中完全对齐时,映射质量为空(即,具有例如零“0”值)。可选地,读数描述符可以包括链性(strandness)描述符,其表示已经测序的链以及执行测序的方向。链性可以由测序机提供的数据组成。可选地,读数描述符可以包括配对链性描述符。实际上,取决于测序技术,读数可以成对提供。在这种情况下,配偶(mate)对应于配对的另一端,通常映射到几千个碱基之外。
[0124]
读数描述符具有一维(1d)结构(一个值针对每个读数)。碱基描述符具有二维(2d)结构(一个值针对每个读数的每个碱基)。碱基描述符包括碱基类型描述符,其定义碱基的类型(即,a、t、c或g)。碱基描述符可以在四个通道上编码(一个通道针对每种碱基类型)。碱基描述符可以包括碱基质量描述符,其描述碱基的标识质量。碱基描述符可以包括参考碱基描述符,其是参考基因组中的对应碱基。碱基描述符可以包括错配描述符,其描述了参考基因组中在碱基与对应碱基之间的差异(例如,基于布尔值)。碱基描述符可以包括插入大小的描述符,并且可替代地或另外地,可以包括删除大小的描述符,该删除大小的描述符可以通过该方法通过比较参考上的每个读数的对齐(包括插入和删除的对齐)来计算。
[0125]
每个数据片段的长度可以是恒定的,并且数据片段可以全部以基因组位置为中心。每个数据片段包括对应于读数的碱基描述符的序列和附加的起始序列78和附加的结束序列79,附加的起始序列78和附加的结束序列79分别位于与读数相对应的碱基描述符的序列的开始和结尾(在图上显示为空白)。附加的起始序列和结束序列的大小变化以对齐参考基因组的上的每个读数的相应的序列。附加的起始序列或结束序列由具有空值的一个或多个碱基描述符的序列组成。长度对应于两个读数的大小(在恒定长度读数的情况下,或者大约是最长读数的两倍),例如,300个碱基。该方法还可能截断数据,并使用较短的长度(低于300个碱基)。恒定长度促进处理(例如,经由卷积)。
[0126]
第3节:变体过滤
[0127]
3.1深度学习模型
[0128]
与基于使用具有数百万个参数且不特定于基因组数据的卷积神经网络(cnn)的最先进的图像分类模型的许多机器学习方法不同,该方法可以实现考虑基因组数据的特异度
的神经网络的架构。这允许减小模型的大小和/或提高准确度。
[0129]
读数是1d结构,并且它们在堆中的表示是任意的。实际上,y坐标(即,在堆积图像中的堆的方向)并不提供信息。在该方法中,神经网络的结构包括与次序无关的算子(例如,总和、平均、最小值、最大值)。因此,相对于基于标准2d cnn的现有技术方法,该方法的神经网络是用于变体识别的改进的解决方案。神经网络具有优化的架构,该架构降低了计算成本并展示高性能。
[0130]
图8示出了用于识别种系变体的神经网络架构的示例。用于种系情况的神经网络由对称函数90组成,该对称函数90被配置为取一组读数指定的数据片段80-81以及堆描述符86作为输入并对其进行处理。每个数据片段85指定相对于基因组位置对齐的相应的读数并且包括读数的碱基描述符的序列(每个碱基描述符在一个或多个通道上编码)。每个数据片段85包括2d结构(即,一个方向针对碱基序列并且一个方向针对与序列的每个碱基相关联的对应碱基描述符)。读数叠加以形成堆,这为该组数据片段提供3d结构80(一个方向针对读数,一个方向针对每个读数的碱基序列并且一个方向针对碱基描述符)。神经网络被配置为输出关于在基因组位置处变体的存在的信息82。
[0131]
用于种系情况的函数90包括若干个卷积层83、84,这些卷积层83、84用于处理每个相应的碱基描述符的序列。每个卷积层应用若干个一维卷积滤波器。函数90包括第一1
×
1卷积层83,该卷积层83用于在预定数量的通道(例如,五个通道)中重新编码碱基描述符。第一卷积层83以均匀和非线性的方式在碱基级别变换信息。第一卷积83允许使输入数据归一化。然后,函数90包括第二lx1卷积层84,该卷积层84用于将每个读数的数据片段从l个位置减少到1个位置,其中l是每个数据片段的大小。第二卷积84输出预定数量的通道,例如,十个通道。函数90包括每个读数的读数描述符81与第二卷积的输出的级联。函数90在第一归约层中输入读数描述符和第二卷积的输出的级联。归约层基于与次序无关的算子(例如,均值和标准差)将与读数描述符级联的第二卷积层的输出与次序无关地折叠为一组特征。归约层将n个读数折叠为一组特征(例如,二十个特征)的一个包。函数90将堆描述符86聚合为该组特征的包。该组对应于读数的单个堆,并且堆描述符指定所述堆。然后,函数90包括被配置用于处理归约层的输出和执行分类的若干个全连接层87。
[0132]
神经网络可能具有5240个可训练参数。机器学习方法可以基于一组超参数对神经网络进行机器学习,该超参数包括学习率(例如,1e-3)、迭代次数(例如,100)、内部验证示例的百分比(例如,10%)、优化算法(例如,adam)、激活函数(例如,mish)和成本函数(例如,加权交叉熵)。
[0133]
图9示出了用于识别体细胞变体的神经网络100的架构的示例。神经网络100取两组读数作为输入:第一组101用于种系变体(即,来自个体(例如,患有癌症疾病的人类患者)的健康细胞的读数),并且第二组101’用于体细胞变体(即,来自同一个体的癌细胞的读数)。神经网络100包括用于处理第一组的第一对称函数和用于处理第二组的第二对称函数。第一函数由子架构102、103、104和105组成,并且第二函数由子架构102、103、104和105’组成。第一函数和第二函数是相同的,即,包括相同的参数值和共享的加权。第一函数和第二函数中的每一个都包括两个卷积层102、103,这两个卷积层102、103用于处理碱基描述符的每个序列。卷积层(第一1x1卷积层102和第二lx1卷积层103)对于第一函数和第二函数是相同的(即,包括用于第一函数和第二函数的共享的加权)。神经网络100将读数描述符与第
二卷积层的输出级联。
[0134]
在级联之后,神经网络100包括与相应的读数描述符级联的第二卷积层的输出的拆分104,以分离对应于第一组的输出和对应于第二组的输出。在拆分之后,第一函数和第二函数中的每一个都包括相应的归约层105、105’,该归约层105、105’用于独立地(例如,连续地或并行地)处理对应于每个组的输出。相应的归约层105、105’中的每一个基于与次序无关的算子将第二卷积层的输出与次序无关地折叠为一组相应的特征。该组特征是第一函数和第二函数的输出。归约层105、105'可以是相同的,即,可以包括相同的与次序无关的算子。
[0135]
然后,神经网络100包括用于聚合第一函数和第二函数的输出(即,对应于第一组的一组特征和对应于第二组的一组特征)的层。例如,聚合层可以级联两组特征,从而形成单组特征106。聚合层可以可选地将堆描述符与每个归约层的输出聚合(例如,将堆描述符与单组特征106级联)。这两个组可以完全对应于同一个读数的堆,并且堆描述符指定所述堆。
[0136]
在聚合层之后,神经网络100包括一个或若干个全连接层107,该一个或若干个全连接层107被配置用于执行分类。若干个全连接层107取聚合层的输出(即,每个归约层的输出和可选的堆描述符的级联)作为输入。与种系情况下讨论的全连接层的不同可能在于输入的大小,在体细胞情况下可能是两倍大。机器学习方法可以例如基于9735个可训练参数对用于体细胞变体的神经网络进行机器学习。
[0137]
3.2性能
[0138]
3.2.1度量
[0139]
现在基于若干个度量呈现神经网络的性能。这些度量衡量阳性类预测得多好。事实上,阴性类是非常多数的,因为位置中的99.9%没有突变(基因组中的位置中的99.9%不包含变体)。在该上下文中,其他度量没有提供信息,因为它们给出了0.999模型预测总是阴性类。
[0140]
在下文中,tp指代真阳性,fp指代假阳性,并且fn指代假阴性。第一度量是精度,其被表述为tp/(tp+fp)。精度衡量用于变体识别的神经网络在预测非假阳性的阳性方面有多好。第二度量是召回,其被表述为tp/(tp+fn)。召回衡量用于变体识别的神经网络在不忘记任何真变体方面有多好。第三度量是f1分数,其被表述为2*精度*召回/(精度+召回)。f1分数是精度和召回的几何均值。第四度量是错误的数量。在基因组规模上,f1分数可能非常接近1.0,以至于很难区分不同的模型性能。在这种情况下,人们可能会使用错误预测的数量(即,错误的数量)来正确区分性能。
[0141]
3.2.2数据集和结果
[0142]
在下文中,神经网络基于公开可用的地面真值数据集进行训练。将神经网络的结果与现有技术变体识别器的列表进行比较。现有技术的变体识别器列表包括gatk4、deepvariant 0.9和cnn分类器efficientnet。
[0143]
现在讨论种系情况的结果。对于种系情况,神经网络和每个现有技术的变体识别器都基于训练集进行训练,该训练集是个体基因组hg001。个体基因组hg001是由genome in a bottle联盟(giab联盟)通过多种技术交叉验证的个体基因组。神经网络和每个现有技术变体识别器的性能是基于验证集计算的,该验证集是个体基因组hg002。基于之前介绍的度
量,性能得到了赞赏。
[0144]
图10示出了针对种系情况的精度、召回和f1分数度量的结果的示例。神经网络的f1分数和现有技术的变体识别器中的每一个的f1分数彼此非常接近。
[0145]
图11示出了针对种系情况的错误数量度量的结果的示例。神经网络的性能接近deepvariant0.9,但只有大约4000个参数,而deepvariant 0.9使用24m个参数。此外,deepvariant0.9已经在300m个示例上进行了训练,而神经网络仅在1m个示例上进行了训练。实际上,当在包括1m个示例的相同数据集上进行训练时,deepvariant 0.9表现出较差的性能。此外,deepvariant 0.9不管理体细胞情况。
[0146]
现在讨论体细胞情况的结果。对于体细胞情况,结果是根据公开可用的体细胞参考标准计算的。结果包括来自colo829癌细胞系和来自同一供体的血细胞系的结果。共有序列(consensus)是在四个独立的illumina平台上进行全基因组测序并使用多个变体识别器后获得的。训练集包含70000个示例。
[0147]
结果还包括来自hcc1143癌细胞系体细胞变体共有序列的结果,该共有序列在两个独立的illumina平台上获得并由现有技术的mutect2、strelka2和lancet变体识别器处理。
[0148]
图12示出了在神经网络与现有技术变体识别器strelka2和lancet之间的针对体细胞情况的比较结果的示例。体细胞测序尤其具有挑战性,并且由于缺乏变体的共有序列地面真值而变得困难。这对于基于学习的方法来说甚至更成问题。然而,结果是基于试图建立共有序列的工作中的地面真值体细胞数据集的近似值。结果表明,神经网络与两种最先进的变体识别器lancet和strelka2相比具有优势。
[0149]
图13示出了系统的示例,其中该系统是客户端计算机系统,例如,用户的工作站。
[0150]
该示例的客户计算机包括连接至内部通信总线1000的中央处理单元(cpu)1010、也连接至总线的随机存取存储器(ram)1070。客户端计算机还被提供有图形处理单元(gpu)1110,该gppu1110与连接到总线的视频随机存取存储器1100相关联。视频ram 1100在本领域中也称为帧缓冲器。大容量存储设备控制器1020管理对大容量存储设备(例如,硬盘驱动器1030)的访问。适合于有形地体现计算机程序指令和数据的大容量存储设备包括所有形式的非易失性存储器,通过示例的方式,包括半导体存储器设备,例如,eprom,eeprom和闪速存储器设备;磁盘,例如,内部硬盘和可移除磁盘;磁光盘;以及cd-rom盘1040。前述内容中的任一项可以通过专门设计的asic(专用集成电路)进行补充或并入该专门设计的asic。网络适配器1050管理对网络1060的访问。客户端计算机还可以包括触觉设备1090,例如,光标控制设备、键盘等。在客户端计算机中使用光标控制设备以允许用户将光标选择性地定位在显示器1080上的任何期望位置。此外,光标控制设备允许用户选择各种命令并输入控制信号。光标控制设备包括用于将控制信号输入到系统的多个信号生成设备。典型地,光标控制设备可以是鼠标,该鼠标的按钮用于生成信号。可替代地或另外地,客户计算机系统可以包括敏感垫和/或敏感屏幕。
[0151]
该计算机程序可以包括可由计算机执行的指令,该指令包括用于使上述系统执行方法的模块。该程序可以记录在包括系统的存储器的任何数据存储介质上。该程序可以例如以数字电子电路装置或计算机硬件、固件、软件或其组合来实现。该程序可以被实现为有形地体现在机器可读存储设备中以由可编程处理器执行的装置,例如,产品。方法步骤可以
这样执行:通过可编程处理器执行指令程序以通过对输入数据进行操作并生成输出来执行该方法的功能。因此,处理器可以是可编程的并且被耦合以从数据存储系统、至少一个输入设备和至少一个输出设备接收数据和指令,以及向数据存储系统、至少一个输入设备和至少一个输出设备发送数据和指令。如果需要,则可以以高级过程编程语言或面向对象的编程语言或汇编或机器语言来实现应用程序。在任何情况下,该语言都可以是编译语言或解释语言。该程序可以是完整的安装程序或更新程序。在任何情况下,程序在系统上的应用都会导致执行方法的指令。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1