具有多级孔电极复合材料GO-C@M(OH)2及制备方法和应用
具有多级孔电极复合材料go-c@m(oh)2及制备方法和应用
技术领域
1.本发明涉及复合电极材料领域,具体涉及一种具有多级孔电极复合材料go-c@m(oh)2及制备方法和应用。
背景技术:
2.随着化石能源的不断消耗,能源危机和环境污染的日趋严重,世界各国都在寻找能够替代化石能源的清洁、高效和可持续的能源。超级电容器是一种不同于传统电容器和蓄电池的新型储能材料,因其具有高功率密度、低使用风险、快速充放电等特点而在最近几年受到广泛关注。而电极材料是决定超级电容器电化学性能关键因素,因此研究具有更高比电容及电化学性能的电极材料是目前超级电容器研究热点。
3.在各种电极材料中,过渡金属氢氧化物由于具有较高的比电容和能量密度,受到研究者的广泛关注,被认为是超级电容器最佳电极材料的候选之一。过渡金属氢氧化物在合成制备时易发生团聚,大部分过渡金属氢氧化物为片状堆积结构,这种堆积结构不利于活性点的暴露,同时单纯金属氢氧化物导电性不好不利于电极电子的传输,这些因素限制了过渡金属氢氧化物在超级电容器电极材料上的应用。
技术实现要素:
4.本发明的目的在于克服上述技术不足,提供一种具有多级孔电极复合材料go-c@m(oh)2及制备方法和应用,解决现有技术中过渡金属氢氧化物易团聚、活性点低以及导电性不好的技术问题。
5.为达到上述技术目的,本发明制备方法的技术方案是:
6.包括以下步骤:
7.(1)将多孔海绵浸泡在go溶液中,得到go修饰多孔海绵,其中多孔海绵与go的质量比为1:(0.1~1);
8.(2)采用水热反应将微孔mofs生长在go修饰多孔海绵上,制备出go-多孔海绵@mofs复合材料,再进行碳化得到go-c@m材料;其中m为mofs中的金属离子;
9.(3)go-c@m材料与含硫化合物进行水热反应制备go-c@mso4,go-c@mso4在碱性溶液中浸泡,得到电极复合材料go-c@m(oh)2。
10.进一步地,海绵为聚氨酯海绵、三聚氰胺海绵、聚苯乙烯海绵、聚乙烯海绵或聚有机硅海绵。
11.进一步地,go溶液的浓度为1~10mg/ml,go溶液中溶剂为水、乙醇、甲醇及dmf中的一种或多种任意比例的混合物。
12.进一步地,步骤(2)中,mofs的原料包括金属源和咪唑类配体,其中go修饰多孔海绵和金属源的质量比为1:(1~5):咪唑类配体和金属源的摩尔比在5以上。
13.进一步地,步骤(2)水热反应中,金属源的浓度为0.05~0.2g/ml;金属源包括硝酸铜、硝酸锌或硝酸钴。
14.进一步地,步骤(2)水热反应是在50~100℃水热反应12~24小时;碳化是在500~800℃碳化2~5小时。
15.进一步地,步骤(3)中,含硫化合物为硫代乙酰胺,水热反应中,go-c@m、硫代乙酰胺和溶剂的比例为0.1g:(0.1~0.3)g:(10~25)ml;步骤(3)中水热反应是在130~150℃反应12~24h。
16.进一步地,步骤(3)中碱性溶液的浓度为1~6mol/l,浸泡时间为24~48h。
17.如上制备方法制得的具有多级孔电极复合材料go-c@m(oh)2。
18.如上具有多级孔电极复合材料go-c@m(oh)2在超级电容器中的应用。
19.与现有技术相比,本发明的有益效果包括:
20.本发明以mofs为金属源,通过在多孔海绵表面以及孔隙内生长mofs,并与go复合,经过高温碳化后得到高分散性的金属纳米粒子,再与含硫化合物水热合成得到氢氧化物纳米粒子。受益于海绵的高开孔率和mofs的微孔结构,所得的go-c@m(oh)2具有多级孔结构并具有很大的比表面积。这种制备方法可以有效的防止氢氧化物纳米粒子的团聚,从而提高电极材料的活性点。此外,氧化石墨烯的加入可以增强复合电极材料的导电性,进而提高go-c@m(oh)2复合材料的电化学性能;本发明go-c/co(oh)2电极在1a/g电流密度下的比容量可达300f/g。
附图说明
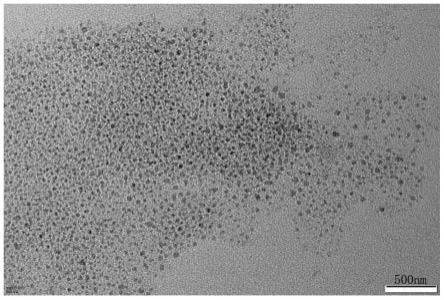
21.图1为本发明中go-c/co(oh)2的tem图;
22.图2为本发明中go-c/co(oh)2电极材料的cv图;
23.图3为本发明中go-c/co(oh)2电极材料的gcd图。
具体实施方式
24.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
25.本发明具有多级孔电极复合材料go-c@m(oh)2的制备方法,包括以下步骤:
26.a、在1~10mg/ml的go溶液中以多孔海绵吸附go,以便得到go修饰的多孔海绵,海绵与氧化石墨烯的质量比为1:0.1~1;
27.b、采用水热反应方法将微孔mofs生长在多孔海绵的内外表面上制备出多孔海绵@mofs复合材料;go修饰的多孔海绵、硝酸钴和2-甲基咪唑的质量比例为1:1~5:4~20;硝酸钴的水溶液的浓度为0.05~0.2g/ml;2-甲基咪唑的浓度为0.2~0.8g/ml;50~100℃水热反应12~24小时;
28.c、在惰性气体下通过高温将多孔海绵@mofs复合材料碳化得go-c@m材料,500~800℃碳化2~5小时;
29.d、通过水热法将go-c@m与硫代乙酰胺进行反应制备go-c@mso4,其中go-c@m、硫代乙酰胺和溶剂(水、乙醇、甲醇及dmf中的一种或多种)的比例为0.1g:0.1~0.3g:10~25ml;130~150℃反应12~24h。
30.e、将go-c@mso4浸泡于浓度为1~6m的过量碱性溶液中24~48h制备go-c@m(oh)2复
合材料。
31.优选的,海绵为聚氨酯海绵、三聚氰胺海绵、聚苯乙烯海绵、聚乙烯海绵及聚有机硅海绵等。
32.优选的,mofs为mof-5、mof-177、zif-5、zif-7、zif-8、zif-11、zif-20、zif-67、zif-68等。
33.优选的,金属离子m为cu
2+
、zn
2+
、co
2+
等。
34.本发明mofs材料中,金属源和咪唑类配体的理论摩尔比为1:1,为了保证金属源的充分反应,采用过量的咪唑类配体,优选金属源5倍以上摩尔量的咪唑类配体,由于所采用的金属源和咪唑类配体均溶于水等溶剂,产物不溶,因此可以得到纯度高的mofs材料;本发明可以根据mofs的具体型号选择金属源以及对应的配体。
35.优选的,go溶液中溶剂为水、乙醇、甲醇及dmf中的一种或多种的混合物。
36.优选的,碱性溶液为氢氧化钠、氢氧化钾溶液。
37.下面通过具体的实施例对本发明做进一步详细说明。
38.实施例1
39.(1)将1cm
×
2cm
×
1cm的三聚氰胺海绵块用乙醇溶液超声波清洗15分钟,60℃干燥1小时,然后放入5mg/ml的氧化石墨烯的水溶液中浸泡至完全吸收,将海绵60℃下干燥,海绵与氧化石墨烯的质量比为1:1,制备得到go修饰的多孔海绵;
40.(2)将上述go修饰的多孔海绵浸泡在含0.1g/ml硝酸钴的水溶液中挤压数次后取出干燥。再放入含有0.4g/ml 2-甲基咪唑的溶液中,在100℃水热反应24小时,反应后固体取出干燥,然后将干燥后的海绵置于碳化炉中600℃碳化2小时,碳化后待冷却至室温后将产物取出并研成粉末得go-c@co材料;其中,go修饰的多孔海绵和硝酸钴的质量比为1:2,硝酸钴和2-甲基咪唑的摩尔比为1:5。
41.(3)称取0.1g硫代乙酰胺与0.1g go-c@co一起溶于10ml去离子水中,超声20min,将混合液放入25ml特氟龙反应釜150℃反应12h。待冷却至室温后取出,用无水乙醇洗涤后干燥,加入到1m过量的氢氧化钠溶液中浸泡24h,过滤,干燥,得到的黑色粉末即为go-c/co(oh)2复合材料。
42.将实施例1制得的go-c/co(oh)2复合材料的粉末、乙炔黑和聚四氟乙烯ptfe以质量比为8∶1∶1混合到一起,用试管手工压成型,然后再放入到钢网中,6m压力压制,形成go-c/co(oh)2电极片;将go-c/co(oh)2复合材料-钢网电极片用作超级电容器电极。
43.电化学性能的测试采用的是科斯特电化学工作站,对材料进行了循环伏安测试(cv),恒流充放电测试(gcd),交流阻抗谱测试(eis)和循环稳定性测试。电解液为3mol/l的koh,在室温下使用三电极体系进行测试,其中以复合材料制成的电极片作为工作电极,铂丝为对电极,银电极为参比电极。
44.gcd中可以得到复合材料在不同电流密度下的放电时间,在根据公式:
45.cs=(i*δt)/(m*δv)
ꢀꢀ
(1)
46.可以算出比电容。其中,cs代表电极材料的比电容(f/g);i代表放电电流(a);δt代表放电时间(s),m代表电极中活性物质的质量(g);δv代表测试体系的电压范围。
47.图1为go-c/co(oh)2复合材料的tem图,从图中可以看出20-30nm粒径co(oh)2粒子均匀分散的go-c上,表明go-c/co(oh)2复合材料能有效阻止co(oh)2粒子的团聚问题。
48.图2为go-c/co(oh)2电极材料的cv图,从图中可以看到cv曲线呈现出明显的氧化还原峰,说明该材料电极主要是由法拉第赝电容构成,电化学反应中电极材料发生了氧化还原反应。这些峰是由于电极表面上的co(oh)2粒子在电化学过程中发生氧化还原化学引起的。
49.图3为go-c/co(oh)2电极材料的gcd图,从图中可以看出恒流充放电曲线高度对称,说明go-c/co(oh)2复合材料具有良好的导电性能和库伦效率。通过公式(1)计算出go-c/co(oh)2电极在1a/g电流密度下的比容量是300f/g,与传统的碳材料和mofs材料相比具有更高的电容性能。
50.实施例2
51.(1)将1cm
×
2cm
×
1cm的聚氨酯海绵块用乙醇溶液超声波清洗15分钟,60℃干燥1小时,然后放入2mg/ml的氧化石墨烯的乙醇溶液中浸泡至完全吸收,将海绵60℃下干燥,海绵与氧化石墨烯的质量比为1:0.2,制备得到go修饰的多孔海绵;
52.(2)将上述go修饰的多孔海绵浸泡在含0.05g/ml硝酸锌的水溶液中挤压数次后取出干燥。再放入含有0.2g/ml 2-甲基咪唑的溶液中,在80℃水热反应12小时,反应后固体取出干燥,然后将干燥后的海绵置于碳化炉中800℃碳化2小时,碳化后待冷却至室温后将产物取出并研成粉末得go-c@zn材料;其中,go修饰的多孔海绵和硝酸锌的质量比为1:1,硝酸锌和2-甲基咪唑的摩尔比为1:6。
53.(3)称取0.1g硫代乙酰胺与0.2g go-c@zn一起溶于12ml去离子水中,超声20min,将混合液放入特氟龙反应釜130℃反应24h。待冷却至室温后取出,用无水乙醇洗涤后干燥,加入到2m过量的氢氧化钠溶液中浸泡26h,过滤,干燥,得到的黑色粉末即为go-c/zn(oh)2复合材料,作为电极时,在1a/g电流密度下的比容量是291f/g。
54.实施例3
55.(1)将1cm
×
2cm
×
1cm的聚苯乙烯海绵块用乙醇溶液超声波清洗15分钟,60℃干燥1小时,然后放入4mg/ml的氧化石墨烯的溶液(溶剂采用甲醇及dmf按体积比1:5的混合液)中浸泡至完全吸收,将海绵60℃下干燥,海绵与氧化石墨烯的质量比为1:0.4,制备得到go修饰的多孔海绵;
56.(2)将上述go修饰的多孔海绵浸泡在含0.15g/ml硝酸锌的水溶液中挤压数次后取出干燥。再放入含有0.5g/ml 2-甲基咪唑的溶液中,在70℃水热反应16小时,反应后固体取出干燥,然后将干燥后的海绵置于碳化炉中700℃碳化2.5小时,碳化后待冷却至室温后将产物取出并研成粉末得go-c@zn材料;其中,go修饰的多孔海绵和硝酸锌的质量比为1:5,硝酸锌和2-甲基咪唑的摩尔比为1:5。
57.(3)称取0.1g硫代乙酰胺与0.3g go-c@zn一起溶于25ml去离子水中,超声20min,将混合液放入特氟龙反应釜140℃反应14h。待冷却至室温后取出,用无水乙醇洗涤后干燥,加入到5m过量的氢氧化钠溶液中浸泡30h,过滤,干燥,得到的黑色粉末即为go-c/zn(oh)2复合材料,作为电极时,在1a/g电流密度下的比容量是289f/g。
58.实施例4
59.(1)将1cm
×
2cm
×
1cm的聚有机硅海绵块用乙醇溶液超声波清洗15分钟,60℃干燥1小时,然后放入8mg/ml的氧化石墨烯的溶液(溶剂采用水及乙醇按体积比1:1的混合液)中浸泡至完全吸收,将海绵60℃下干燥,海绵与氧化石墨烯的质量比为1:0.8,制备得到go修
饰的多孔海绵;
60.(2)将上述go修饰的多孔海绵浸泡在含0.2g/ml硝酸铜的水溶液中挤压数次后取出干燥。再放入含有0.8g/ml 2-甲基咪唑的溶液中,在50℃水热反应20小时,反应后固体取出干燥,然后将干燥后的海绵置于碳化炉中500℃碳化3小时,碳化后待冷却至室温后将产物取出并研成粉末得go-c@cu材料;其中,go修饰的多孔海绵和硝酸铜的质量比为1:4,硝酸铜和2-甲基咪唑的摩尔比为1:5。
61.(3)称取0.1g硫代乙酰胺与0.2g go-c@cu一起溶于20ml去离子水中,超声20min,将混合液放入特氟龙反应釜135℃反应16h。待冷却至室温后取出,用无水乙醇洗涤后干燥,加入到4m过量的氢氧化钠溶液中浸泡28h,过滤,干燥,得到的黑色粉末即为go-c/cu(oh)2复合材料,作为电极时,在1a/g电流密度下的比容量是279f/g。
62.对比例1
63.将海绵替换成同样尺寸的聚氨酯塑料块,其他条件同实施例2。
64.所得材料在1a/g电流密度下的比容量为30f/g,由此说明本发明采用海绵这种多孔材料,有效提高了mofs的生长面积,所得最终材料的比表面积和活性位点增大,利于提高材料的电化学性能。
65.本发明以三聚氰胺、聚氨酯等海绵为基底、金属有机框架化合物(mofs)为金属离子源与石墨烯(go)复合,成功制备了go-c@m(oh)2复合材料。利用海绵开孔率高的特点,在海绵的外表面以及孔隙内生长mofs,增大复合材料的比表面积,受益于海绵的高开孔率,所得复合材料具有多级孔结构和很大的比表面积,同时这种方法有效的防止了氢氧化物纳米粒子的团聚,从而提高电极材料的活性点。此外,石墨烯的加入可以增强复合电极材料的导电性,进而提高go-c@m(oh)2复合材料的电化学性能。因此,本发明所得到的go-c@m(oh)2复合材料具有独特的多级孔结构,表现出较优秀的电化学性能。
66.以上所述本发明的具体实施方式,并不构成对本发明保护范围的限定。任何根据本发明的技术构思所做出的各种其他相应的改变与变形,均应包含在本发明权利要求的保护范围内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1