一种合金催化剂及其制备方法和应用与流程
1.本发明属于催化剂技术领域,涉及一种合金催化剂及其制备方法和应用。
背景技术:
2.随着经济的快速发展,能源与环境问题成为人类社会日益关注的现实问题。质子交换膜燃料电池(pemfcs)以其高能量密度、高能源转换效率、低/零排放、高可靠性等特点被视为最具潜力替代煤、石油、天然气等化石能源的新型能源装置之一,未来可被广泛应用在交通、备份电源、固定电站、手持电子设备等领域。然而,阴极氧还原(orr)缓慢的动力学过程以及高昂的铂基(pt)贵金属催化剂等因素极大地阻碍了pemfcs的商业化发展。
3.在过去的二十年里,燃料电池氧还原电催化剂的发展已经取得了巨大的成就,引入非贵金属的多种类型的pt基合金pt-m(m=fe、ni、co、cr etc.)被广泛研究。在这些合金催化剂中,ptco合金催化剂显示出相对更高的催化活性,备受业界关注,甚至连最具代表性的丰田mirai也是采用此类催化剂。然而,因为co相较于fe、ni更难还原的属性,目前市场上所使用的ptco催化剂普遍存在co含量低的特点。例如duan.et al.等人(advanced energy materials,2022,12(13):2103144.)以及wang.et al.等人(acs applied materials&interfaces,2020,12(27):30381-30389.)报道的一种基于微波辅助的乙二醇还原方法,仅仅有不到1%的co离子被还原,造成了前驱体的大量浪费以及环境污染。此外,在燃料电池恶劣的反应条件下,过渡金属co容易溶解、团聚,从而降低了电化学活性面积,最终表现出较差的催化活性和耐久性。
4.cn101359744a公开了一种电沉积制备超低铂催化电极的方法,在水溶液中通过四步电沉积法将高分散的过渡金属m(如:cu、co、ni等)纳米粒子沉积在多孔碳电极(pce)上,然后将所得m/pce电极浸入到氮气保护的铂盐溶液中,通过置换反应得到高分散的碳载超低铂含量催化电极。该方法充分发挥脉冲电沉积瞬时电流高的优势,克服了两步脉冲电沉积过程中由双电层电容充放电所导致的m核晶粒和m@pt晶粒过大、尺寸难以控制的弊端。但此制备方法,仍然存在过程繁琐,影响因素过多,核与壳的金属质量比难以控制等一系列问题,而且铂的用量没有降低。
5.cn100398211a介绍了一种用化学置换法制备核壳催化剂的一种方法,该方法是将非贵金属盐配制成溶液,加入一定量的表面活性剂,然后向混合溶液中加入过量的还原剂,制成非贵金属的纳米金属溶液,再向非贵金属的纳米金属溶液中加入贵金属盐溶液进行化学置换,得到贵金属包裹在非贵金属纳米颗粒表面的核壳结构催化剂溶液,得到非担载型核壳催化剂,最后在非担载型核壳催化剂溶液中加入碳载体进行吸附,得到担载型核壳催化剂。该方法的优点是操作过程简单,制备成本低,但催化剂粒径偏大,而且催化剂仅靠吸附作用与碳载体连接,很容易在电化学反应过程中脱落,从而降低催化效率。
6.cn114335580a报道了一种利用5%h2/ar混合气在600℃高温下4h还原pt离子、co离子的方法。该方法的优点是可以同步还原pt离子和co离子,但其制备的ptco合金呈无序结构,氧化还原反应极化曲线的半波电位也仅仅约0.9v,与使用纯氩气相比,催化剂的活性
不仅没有被明显提升,还增加了实验成本与涉氢的危险性。
7.因此,如何提供一种成本低,方法简单,且催化活性高,耐久性好的铂合金orr电催化剂,是亟待解决的技术问题。
技术实现要素:
8.本发明的目的在于提供一种合金催化剂及其制备方法和应用。本发明通过简单的搅拌以及热处理两步工艺,即可得到活性与耐久性兼具的高性能pt-m/c合金催化剂,适用于多种过渡金属m,且对于过渡金属的还原转化率高,还可以得到大批量的催化剂产品,而且没有使用任何有毒、有害的溶剂,绿色环保,省去了去除有害吸附基团的步骤,方法简单高效,且制备过程不依赖特殊的实验设备,降低了成本。
9.为达到此发明目的,本发明采用以下技术方案:
10.第一方面,本发明提供一种合金催化剂的制备方法,所述制备方法包括以下步骤:
11.(1)将铂碳混合源、过渡金属源和分散剂混合分散,得到混合溶液;
12.(2)将步骤(1)所述混合溶液进行恒温搅拌,直至混合溶液达到粘稠状态,干燥,得到干燥后的物质;
13.(3)将步骤(2)所述干燥后的物质在保护性气氛下进行煅烧,得到所述合金催化剂。
14.本发明步骤(2)中的粘稠状态为混合溶液搅拌至无可流动液体的状态。
15.本发明通过简单的搅拌以及热处理两步工艺,即可得到活性与耐久性兼具的高性能pt-m/c合金催化剂,适用于多种过渡金属m,且对于过渡金属的还原转化率高,还可以得到大批量的催化剂产品,而且没有使用任何有毒、有害的溶剂,绿色环保,省去了去除有害吸附基团的步骤,方法简单高效,且制备过程不依赖特殊的实验设备,降低了成本。
16.本发明中,在步骤(2)先进行恒温搅拌,可以使得金属离子均匀地吸附在碳载体上,在此基础上,无需还原性气氛,直接高温还原金属离子并与pt进行合金化,即可得到活性与耐久性兼具的高性能pt-m/c合金催化剂。
17.本发明中,超声分散后直接干燥,会导致离子吸附不均匀,进而导致制备的pt-m/c合金催化剂的纳米粒子在碳载体上分散不均匀,粒径离散大;如果在还原性气氛下进行高温煅烧处理,比如h2/ar混合气,不仅增加实验成本,而且还会涉及工艺安全问题,且对制备的pt-m/c合金催化剂性能没有明显提升。
18.优选地,步骤(1)所述铂碳混合源包括铂碳催化剂和/或铂源与碳载体的混合源。
19.本发明中,可以以商售的铂碳催化剂为原料,也可以以铂源与碳载体作为原料。
20.优选地,所述铂源包括六水氯铂酸和/或乙酰丙酮铂。
21.优选地,所述碳载体包括导电炭黑。
22.优选地,步骤(1)所述过渡金属源包括钴源、铁源或镍源中的任意一种或至少两种的组合,优选为钴源。
23.优选地,所述钴源包括六水硝酸钴、六水氯化钴或乙酰丙酮钴中的任意一种或至少两种的组合,优选为六水硝酸钴。
24.优选地,步骤(1)所述分散剂包括超纯水。
25.优选地,步骤(1)中,铂碳混合源中的铂的摩尔量与过渡金属源中过渡金属的摩尔
量之比为1:10~10:1,例如1:10、1:5、1:1、2:1、3:1、5:1、8:1或10:1等。
26.优选地,铂碳混合源与过渡金属源的总质量与分散剂的质量体积比为5~20mg/ml,例如5mg/ml、6mg/ml、7mg/ml、8mg/ml、9mg/ml、10mg/ml、11mg/ml、12mg/ml、13mg/ml、14mg/ml、15mg/ml、16mg/ml、17mg/ml、18mg/ml、19mg/ml或20mg/ml等。
27.本发明中,铂碳混合源与过渡金属源的总质量与分散剂的质量体积比过小,催化剂制备过程中会消耗更多的时间,导致制备效率降低;而质量体积比过大,又会导致纳米粒子在碳载体上分散不均匀,使得制备的催化剂性能降低。
28.优选地,步骤(1)所述混合分散的方法为超声分散。
29.优选地,步骤(2)所述恒温搅拌的温度为70~100℃,例如70℃、75℃、80℃、85℃、88℃、90℃、93℃、95℃或100℃等。
30.本发明中,恒温搅拌的温度过低,催化剂制备过程中同样会消耗更多的时间,导致制备效率降低;而温度如果过高,不仅增加了实验风险,而且因为加快分散剂的蒸发导致纳米粒子在碳载体上分散不均匀,使得制备的催化剂性能降低。
31.优选地,步骤(2)所述恒温搅拌的搅拌转速为300~600rpm,例如300rpm、350rpm、400rpm、450rpm、500rpm、550rpm或600rpm等。
32.优选地,步骤(2)所述恒温搅拌的方法包括油浴搅拌和/或沙浴搅拌。
33.优选地,步骤(2)所述干燥的方法为真空干燥。
34.本发明中,选用真空干燥,不仅有利于样品的快速干燥,而且降低了催化剂中pt被氧化的风险,而如果选用鼓风干燥,不仅可能会吹散催化剂粉末,而且会增加催化剂中pt被氧化的风险,进而导致催化剂的性能降低。
35.优选地,所述真空干燥的温度为50~70℃,例如50℃、55℃、60℃、65℃或70℃等。
36.优选地,所述真空干燥的时间为6~12h,例如6h、7h、8h、9h、10h、11h或12h等。
37.优选地,步骤(3)所述煅烧前,将步骤(2)所述干燥后的物质进行研磨。
38.优选地,所述研磨的时间为5~10min,例如5min、6min、7min、8min、9min或10min等。
39.优选地,所述煅烧的温度为800~1000℃,例如800℃、830℃、850℃、880℃、900℃、930℃、950℃、980℃或1000℃等。
40.本发明中,煅烧温度过低,不利于过渡金属离子的完全还原,造成投料的浪费,而煅烧温度过高,又会导致催化剂纳米粒子的严重团聚,使得催化剂中活性成分的利用率降低,同时也会极大地降低催化剂的性能。
41.优选地,所述煅烧的时间为1~3h,例如1h、2h或3h等。
42.优选地,所述保护性气氛为氩气气氛。
43.优选地,对步骤(3)所述煅烧后的产物依次进行洗涤、真空干燥和研磨。
44.作为优选的技术方案,所述制备方法包括以下步骤:
45.(1)将铂碳混合源、钴源和分散剂超声分散,其中,铂碳混合源中的铂的摩尔量与钴源中钴的摩尔量之比为1:10~10:1,铂碳混合源与钴源的总质量与分散剂的质量体积比为5~20mg/ml,得到混合溶液;
46.(2)将步骤(1)所述混合溶液以300~600rpm的搅拌转速进行70~100℃下的恒温搅拌,直至混合溶液达到粘稠状态,50~70℃下真空干燥6~12h,得到干燥后的物质;
47.(3)将步骤(2)所述干燥后的物质研磨5~10min,然后在氩气气氛下以800~1000℃的温度煅烧1~3h,对煅烧后的产物依次进行洗涤、真空干燥和研磨,得到所述合金催化剂。
48.第二方面,本发明提供一种合金催化剂,所述合金催化剂由如第一方面所述的合金催化剂的制备方法制备得到,所述合金催化剂的结构通式为pt-m/c,其中,m为过渡金属。
49.优选地,所述pt-m/c中,m包括co、fe或ni中的任意一种或至少两种的组合,优选为co。
50.第三方面,本发明还提供一种合金催化剂的应用,所述应用包括将如第二方面所述的合金催化剂用于燃料电池阴极氧还原反应中。
51.相对于现有技术,本发明具有以下有益效果:
52.本发明通过简单的搅拌以及热处理两步工艺,即可得到活性与耐久性兼具的高性能pt-m/c合金催化剂,适用于多种过渡金属m,且对于过渡金属的还原转化率高,还可以得到大批量的催化剂产品,且得到的合金催化剂的产量高,不仅可以得到毫克级别的催化剂材料,还可得到克级别的催化剂材料,而且没有使用任何有毒、有害的溶剂,绿色环保,省去了去除有害吸附基团的步骤,方法简单高效,且制备过程不依赖特殊的实验设备,降低了成本。本发明提供的合金催化剂,半波电位可达0.82v以上,且其质量活性(a/mg
pt
@0.9v)可达0.16以上,进一步地采用超声分散手段,同时调控恒温搅拌的温度与煅烧的温度,本发明提供的合金催化剂半波电位可达0.87v以上,且其质量活性(a/mg
pt
@0.9v)可达0.21以上,且其耐久性测试(10000圈)的活性衰减率低至3.7%以下。
附图说明
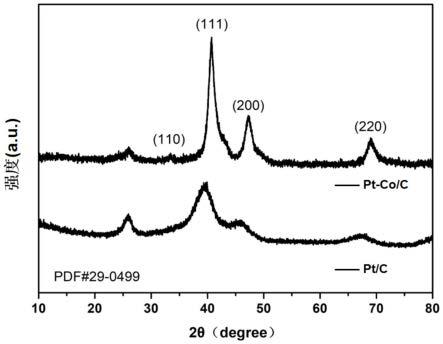
53.图1为实施例1与对比例1提供的合金催化剂的xrd对比图。
54.图2为实施例1与对比例1提供的合金催化剂的电化学测试orr曲线对比图。
55.图3为实施例1与对比例1提供的合金催化剂的电化学测试中的半波电位以及质量活性对比图。
具体实施方式
56.下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
57.实施例1
58.本实施例提供一种pt-co/c合金催化剂材料,所述制备方法如下:
59.(1)称取80mg的co(no3)2·
6h2o固体(钴的摩尔量为0.275mmol)、142mg的h2ptcl6·
6h2o(铂的摩尔量0.275mmol)固体以及63mg的导电炭黑(型号xc-72)载体溶于30ml超纯水中,然后置于超声池中超声10min使其混合均匀,得到铂钴盐/炭黑混合溶液;
60.(2)使用导热介质为二甲基硅油的油浴加热方式,加热至80℃后,将步骤(1)中的混合溶液置于油浴环境中,同时设置磁子搅拌速度为400rpm,等待混合溶液中溶剂蒸干至粘稠状态后转移至真空烘箱中进行真空干燥,温度设置为50℃,时间设置为8h,加热结束后关闭温箱,待恢复至室温25℃后取出;
61.(3)将步骤(2)中所得固体在玛瑙研钵中手工研磨5min至肉眼可见均匀,然后转移
至管式炉中进行煅烧处理,煅烧温度设置为900℃,时间设置为2h,等待程序结束,样品温度降至室温25℃后取出;
62.(4)将步骤(3)中所得固体依次用乙醇、超纯水分别洗涤3次后,转移样品至真空烘箱中进行真空干燥,温度设置为50℃,时间设置为8h,加热结束后关闭温箱,待恢复至室温25℃后取出,最后将样品在玛瑙研钵中手工研磨5min至肉眼可见均匀,即可得到所述pt-co/c合金催化剂材料。
63.实施例2
64.本实施例与实施例1的区别为,本实施例步骤(1)为:称取80mg的co(no3)2·
6h2o固体(钴的摩尔量为0.275mmol)以及120mg的商业化pt/c催化剂(铂的占比为44.7wt%,摩尔量0.275mmol),溶于30ml超纯水中,然后置于超声池中超声10min使其混合均匀,得到钴盐-pt/c混合溶液。
65.其余制备方法与参数与实施例1保持一致。
66.实施例3
67.本实施例与实施例1的区别为,该实施例为催化剂的克级放大制备工艺,本实施例步骤(1)为:称取233mg的co(no3)2·
6h2o固体(钴的摩尔量为0.8mmol)以及1048mg的商业化pt/c催化剂(铂的占比为44.7wt%,摩尔量2.4mmol),溶于90ml超纯水中,然后置于超声池中超声10min使其混合均匀,得到钴盐-pt/c混合溶液。
68.其余制备方法与参数与实施例1保持一致。
69.实施例4
70.本实施例与实施例1的区别为,本实施例步骤(1)中的超纯水的体积为15ml。
71.其余制备方法与参数与实施例1保持一致。
72.实施例5
73.本实施例与实施例1的区别为,本实施例步骤(2)中油浴的温度为100℃。
74.其余制备方法与参数与实施例1保持一致。
75.实施例6
76.本实施例与实施例1的区别为,本实施例步骤(3)中煅烧的温度为800℃。
77.其余制备方法与参数与实施例1保持一致。
78.实施例7
79.本实施例与实施例1的区别为,本实施例步骤(3)中煅烧的温度为1000℃。
80.其余制备方法与参数与实施例1保持一致。
81.实施例8
82.本实施例与实施例1的区别为,本实施例步骤(1)中直接将固体物质分散于水中,不进行超声分散。
83.其余制备方法与参数与实施例1保持一致。
84.实施例9
85.本实施例与实施例1的区别为,本实施例步骤(2)中油浴的温度为120℃。
86.其余制备方法与参数与实施例1保持一致。
87.实施例10
88.本实施例与实施例1的区别为,本实施例步骤(3)中煅烧的温度为1100℃。
89.其余制备方法与参数与实施例1保持一致。
90.实施例11
91.本实施例与实施例1的区别为,本实施例步骤(3)中煅烧的温度为700℃。
92.其余制备方法与参数与实施例1保持一致。
93.实施例12
94.本实施例提供pt-ni/c合金催化剂材料,所述制备方法如下:
95.本实施例与实施例1的区别为,本实施例步骤(1)为:称取87.2mg的ni(no3)2·
6h2o固体(镍的摩尔量为0.3mmol)以及130.9mg的商业化pt/c催化剂(铂的占比为44.7wt%,摩尔量0.3mmol),溶于30ml超纯水中,然后置于超声池中超声10min使其混合均匀,得到镍盐-pt/c混合溶液。
96.其余制备方法与参数与实施例1保持一致。
97.对比例1
98.本对比例提供一种市售铂碳催化剂(铂的质量占比为46.2%)。
99.图1示出了实施例1与对比例1提供的合金催化剂的xrd对比图,由图1可知,实施例1制备的pt-co/c合金催化剂属于面心立方(fcc)结构(pdf#29-0499),与对比例1中的商用pt/c催化剂相比,实施例1所制备pt-co/c合金催化剂的(110)、(111)、(200)、(220)衍射峰均向右偏移,且处在pt(pdf#04-0802)和co(pdf#15-0806)对应的衍射峰之间,说明co原子已经成功被引入pt的晶格中,最终形成copt3合金结构,通过粒径分析,所制备的ptco/c合金催化剂中pt-co合金纳米颗粒的粒径为4~7nm。
100.图2示出了实施例1与对比例1提供的合金催化剂的电化学测试orr曲线对比图,由图2可知,在转转圆盘电极(rde)上相同铂载量的情况下,实施例1中所制备的pt-co/c合金催化剂的半波电位明显高于对比例1提供的商业pt/c催化剂。
101.图3为实施例1与对比例1提供的合金催化剂的电化学测试中的半波电位以及质量活性对比图,实施例1中所制备的pt-co/c合金催化剂的半波电位为0.9v,商业pt/c催化剂的半波电位为0.88v;此外,经过归一化处理转化为质量活性后,实施例1中所制备的pt-co/c合金催化剂的质量活性为0.27a/mg
pt
@0.9v,比商业pt/c催化剂0.13a/mg
pt@0.9v
的2倍还要多,展示出了优异的orr催化活性。
102.对比例2
103.本对比例与实施例1的区别为,本对比例步骤(2)中不进行油浴蒸干操作,具体为在步骤(1)中的超声混合结束后,直接将混合物置于干燥箱中进行真空干燥。
104.其余制备方法与参数与实施例1保持一致。
105.对实施例1-12与对比例1-2提供的合金催化剂进行icp-oes元素分析测试,其结果如表1所示。
106.表1
[0107][0108]
从实施例1-12的数据结果可知,通过本发明提供的制备方法,过渡金属实现了较为完全的还原。
[0109]
将实施例1-12与对比例1-2提供的合金催化剂墨水滴在经氧化铝粉末抛光的玻碳电极上作为工作电极,以碳棒或者铂片作为对电极,以可逆氢电极作为参比电极,构建三电极体系,于0.1m的高氯酸溶液中进行氧还原电催化活性测试。电化学活性测试结果如表2所示。
[0110]
表2
[0111][0112][0113]
进一步地,对实施例1和对比例1提供的合金催化剂进行和耐久性测试,其中耐久性测试的电压扫描范围为0.6-0.95v(vs.rhe),扫描圈数为10000圈。电化学耐久性测试结果如表3所示。
[0114]
表3
[0115][0116]
结合表2和表3的数据结果可知,
[0117]
从实施例1与实施例8、实施例9的数据结果可知,不进行超声分散或者油浴温度太高,纳米粒子在碳载体上会分散不均匀,导致制备的催化剂性能较差。
[0118]
从实施例1与实施例10和实施例11的数据结果可知,煅烧温度过高,会导致纳米颗粒团聚严重,而煅烧温度过低,又难以实现过渡金属离子的完全还原,最终导致前驱体过渡金属源的浪费以及较差的催化剂的性能。
[0119]
从实施例1与对比例1的数据结果可知,本发明制备得到的合金催化剂,与商业化的pt/c催化剂相比,催化活性更高,且耐久性更好。
[0120]
从实施例1与对比例2的数据结果可知,实施例1中的步骤(2)制备均匀粘稠状混合物再进行干燥的过程中非常重要,否则ptco合金纳米颗粒会在碳载体上发生严重团聚且分散不均匀,最终导致活性极差。
[0121]
综上所述,本发明通过简单的搅拌以及热处理两步工艺,即可得到活性与耐久性兼具的高性能pt-m/c合金催化剂,适用于多种过渡金属m,且对于过渡金属的还原转化率高,还可以得到大批量的催化剂产品,且得到的合金催化剂的产量高,不仅可以得到毫克级别的催化剂材料,还可得到克级别的催化剂材料,而且没有使用任何有毒、有害的溶剂,绿色环保,省去了去除有害吸附基团的步骤,方法简单高效,且制备过程不依赖特殊的实验设备,降低了成本。本发明提供的合金催化剂,半波电位可达0.82v以上,且其质量活性(a/mg
pt
@0.9v)可达0.16以上,进一步地采用超声分散手段,同时调控恒温搅拌的温度与煅烧的温度,本发明提供的合金催化剂半波电位可达0.87v以上,且其质量活性(a/mg
pt
@0.9v)可达0.21以上,且其耐久性测试(10000圈)的活性衰减率低至3.7%以下。
[0122]
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1