一种DNA自索引区间解压缩方法
一种dna自索引区间解压缩方法
技术领域
1.本发明涉及dna压缩数据的解压缩技术领域,具体涉及一种dna自索引区间解压缩方法。
背景技术:
2.随着dna测序技术的发展,生物医学研究面临着如何存储和传输dna数据的问题。对dna数据进行压缩后,然后再进行解压缩的技术成为其中解决问题的重要方法之一。
3.lyzip工具基于tpbwt算法进行数据压缩得到短读测序数据后,采用现有的解压缩算法只能实现全局的、静态的解压缩。现有的解压缩算法虽然能够实现dna数据的解压缩,但是需要的解压缩时间较长、且解压缩后的数据需要的存储空间也较大,因此,提出一种减少解压缩时间和存储空间的方法是十分必要的。
技术实现要素:
4.本发明的目的是为解决现有的解压缩算法需要的解压缩时间长,且解压缩后的数据需要的存储空间大的问题,而提出了一种dna自索引区间解压缩方法。
5.本发明为解决上述技术问题采取的技术方案是:一种dna自索引区间解压缩方法,所述方法具体包括以下步骤:
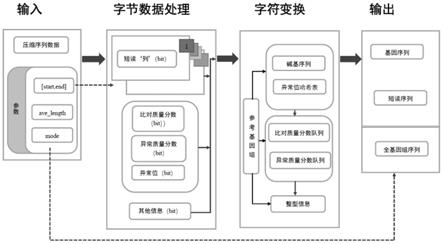
6.步骤一、输入待解压缩的序列数据文件,并配置索引区间参数和解压缩输出模式参数;
7.步骤二、根据索引区间参数,确定出待解压缩的序列数据文件中需要解压缩的区间范围;
8.步骤三、根据待解压缩序列数据文件的头文件信息,确定出需要解压缩区间范围内的测序短读碱基比特信息,比对到参考基因组上碱基的测序质量分数比特信息,无法比对到参考基因组上单核苷酸变异的测序质量分数比特信息,单核苷酸变异、插入删除变异、结构变异比特信息以及表示位置、长度比特信息的具体位置;
9.步骤四、分别对测序短读碱基比特信息,比对到参考基因组上碱基的测序质量分数比特信息,无法比对到参考基因组上单核苷酸变异的测序质量分数比特信息,单核苷酸变异、插入删除变异、结构变异比特信息以及表示位置、长度比特信息进行字节变换处理后,获得字节变换后的压缩文件;
10.步骤五、对字节变换后的压缩文件进行字符变换,还原出每一列所包含的信息;
11.步骤六、将还原出的每一列所包含的信息进行存储后,并按照步骤一配置的解压缩输出模式参数进行输出。
12.本发明的有益效果是:本发明提出了一种dna自索引区间解压缩方法,本发明的自索引区间解压缩算法可以根据需求来选取解压缩的范围,相对于全局静态tpbwt解压缩算法来说,很大程度的降低了解压缩时间,同时也降低了解压缩数据的存储空间。相对于传统解压缩算法,该算法更加灵活能够依据不同需求,解压缩出不同含义的数据,适用性更强。
附图说明
13.图1为自索引区间解压缩方法的流程图;
14.图2为dtpbwt算法的流程图。
具体实施方式
15.具体实施方式一:结合图1说明本实施方式。本实施方式所述的一种dna自索引区间解压缩方法,所述方法具体通过以下步骤实现:
16.步骤一、输入待解压缩的序列数据文件,并配置索引区间参数([start,end])和解压缩输出模式参数(mode);
[0017]
步骤二、根据索引区间参数,确定出待解压缩的序列数据文件中需要解压缩的区间范围;
[0018]
步骤三、根据待解压缩序列数据文件的头文件信息,确定出需要解压缩区间范围内的测序短读碱基比特信息(简称短读“列”),能够比对到参考基因组上碱基的测序质量分数比特信息(简称比对质量分数),无法比对到参考基因组上单核苷酸变异的测序质量分数比特信息(简称异常质量分数),单核苷酸变异、插入删除变异、结构变异比特信息(简称异常值)以及表示位置、长度比特信息(简称其他信息)的具体位置;
[0019]
步骤四、分别对测序短读碱基比特信息,比对到参考基因组上碱基的测序质量分数比特信息,无法比对到参考基因组上单核苷酸变异的测序质量分数比特信息,单核苷酸变异、插入删除变异、结构变异比特信息以及表示位置、长度比特信息进行字节变换处理后,获得字节变换后的压缩文件;
[0020]
步骤五、在引入参考基因组的基础上,对字节变换后的压缩文件进行字符变换,还原出每一列所包含的信息;
[0021]
步骤六、将还原出的每一列所包含的信息进行存储后,并按照步骤一配置的解压缩输出模式参数进行输出。
[0022]
本实施方式还可以配置序列长度保留参数(ave_length)作为可选参数,序列长度保留参数是一个长度信息与步骤二确定的序列需要解压缩长度信息进行比较。如果则扩大解压缩区间,直至解压缩出一条完整的序列。如果则缩小解压缩区间或者直接舍弃,不对序列进行长度扩展。
[0023]
输入的压缩测序数据根据不同的需求,解压缩之后将会得到不同用途的碱基序列数据。根据预设的参数可以分成以下三种情况。
[0024]
1)解压缩索引区间远大于平均序列长度。压缩文件是全基因组拼接序列数据时,解压缩过程相当于还原整个全基因组序列。依据提供的索引区间最大化的解压缩数据区间。在解压缩过程中依据序列之间的overlap重组出全部或者是部分全基因序列数据。
[0025]
当起始start索引位置和终止end索引位置都恰好是overlap区间位置,则保留所有索引区间内的短读序列,并且根据overlap信息将序列进行拼接。
[0026]
当起始start索引位置或终止end索引位置处于正常测序短读区间内,则自动延长索引区间范围,直至到达overlap标志区间终止。将[start’,end]或[start,end’]内序列按照overlap信息完成解压缩组装。
[0027]
当起始start索引位置和终止位置将全部overlap序列包含在内,即可以实现全基因组的解压缩和组装。
[0028]
2)解压缩索引区间远小于平均序列长度。当提供的压缩文件中包含明确的基因位点信息时,可以根据提供的基因位点信息完成对于特定基因序列文件的解压缩操作。输入文件为包含基因位点的测序短读序列,此时特定位点的基因是一个区间位置,该区间位置就是解压缩过程中所需要输入的索引区间位置信息。基因可能位于一条完整的短读内,也可以被多个短读分别存储。解压缩过程中不再去解压缩一条完整的测序短读,而是将输入数据看成数据块,只提取所需要区间的信息。解压缩得到的基因数据可能是多条测序序列的组合信息,但并不影响最终解压缩的效果。解压缩索引区间内包含多列组合数据,每一列有可能是由同一种字符组成也可能是由不同字符组成。字符相同时将一列存储为一个字符;字符不同时分别进行存储。通过上述操作可以将等位基因进行解压缩存储,不会造成变异信息的损失。
[0029]
3)解压缩索引区间长度近似等于平均序列长度。对于解压缩数据不做特殊要求,即按照一条短读测序数据进行解压缩处理。短读序列的解压缩数据由于索引区间范围较大,对于计算机识别具体需求较为困难,可以设定模式参数指定将压缩文件按照完整短读序列进行解压缩处理。通常对于给定的索引区间范围,会出现以下三种测序短读解压缩处理状况。索引范围区间包含一条完整的read,处理较为简单,直接将整条read解压缩存储即可。当索引区间只能包含部分短读序列,可以设定不同的参数指定短读的保留情况。根据需求,可以设定参数—ave_length指定解压缩过程中将索引区间范围的序列进行保留。此时会一定程度的延长索引范围,从而保证能够解压缩出所有短读序列的全部信息而非部分信息,也可以设定参数(ave_length)对于已解压缩片段长度小于ave_length值的序列不予保存。
[0030]
具体实施方式二:本实施方式是对具体实施方式一的进一步具体说明,所述解压缩输出模式参数决定解压缩输出的数据类型。
[0031]
具体实施方式三:本实施方式是对具体实施方式二的进一步具体说明,所述解压缩输出模式参数设置为1时,则解压缩输出的数据类型为基因序列,当解压缩输出模式参数设置为2时,则解压缩输出的数据类型为短读序列,当解压缩输出模式参数设置为3时,则解压缩输出的数据类型为全基因组序列。
[0032]
默认情况下为0,按照短读序列情况进行解压缩。
[0033]
具体实施方式四:本实施方式是对具体实施方式三的进一步具体说明,所述待解压缩序列数据文件的头文件信息包括100位比特信息。
[0034]
每20位表示一个整型位置信息,20位比特值对应着不同数据类型在压缩文件中的比特长度信息。根据这一长度信息可以快速划分,将压缩文件分成完整的5部分。
[0035]
具体实施方式五:本实施方式是对具体实施方式四的进一步具体说明,所述步骤四中,对测序短读碱基比特信息进行字节变换处理的方式为:
[0036]
对测序短读碱基比特信息进行游程解码,将原始的测序短读碱基比特信息还原为整型的{0,1}数据,得到碱基序列。
[0037]
具体实施方式六:本实施方式是对具体实施方式五的进一步具体说明,所述步骤四中,对比对到参考基因组上碱基的测序质量分数比特信息,无法比对到参考基因组上单
核苷酸变异的测序质量分数比特信息以及单核苷酸变异、插入删除变异、结构变异比特信息进行字节变换处理的方式为:
[0038]
采用霍夫曼解码,将比对到参考基因组上碱基的测序质量分数比特信息,无法比对到参考基因组上单核苷酸变异的测序质量分数比特信息以及单核苷酸变异、插入删除变异、结构变异比特信息还原为字符型数据,得到正常比对质量分数队列、异常质量分数队列以及异常值哈希表。
[0039]
其中,单核苷酸变异、插入删除变异、结构变异的比特信息还原为{a,t,c,g}字符,而能够比对到参考基因组上碱基的测序质量分数的比特信息以及无法比对到参考基因组上单核苷酸变异的测序质量分数的比特信息还原为小于40的字符型数值。
[0040]
具体实施方式七:本实施方式是对具体实施方式六的进一步具体说明,所述步骤四中,对表示位置、长度比特信息进行字节变换处理的方式为:
[0041]
采用算术编码压缩对表示位置、长度比特信息进行解码,得到整型信息。
[0042]
具体实施方式八:本实施方式是对具体实施方式七的进一步具体说明,所述步骤五的具体过程为:
[0043]
步骤五一、对字节变换后的压缩文件的每一列进行dtpbwt解压缩变换,改变每一列中的碱基序列和异常值哈希表的异常值顺序;
[0044]
步骤五二、对正常比对质量分数队列进行解压缩,解压缩策略是有损均值化分箱处理的逆过程;
[0045]
正常质量分数同一位点下质量分数较为接近。如果出现差别较大离群点,可以认为是测序异常,将该值忽略。所以对于正常质量分数采取有损均值化分箱处理,对应的解压缩策略便是该策略的逆过程;
[0046]
步骤五三、对异常质量分数队列进行解压缩,解压缩策略是有损离散化分箱处理的逆过程;
[0047]
同时引入了随机变量,使得原有的解压缩质量分数均值有一定的偏差。异常质量分数代表每个异常碱基的测序质量。通常来说每一列的异常值都不相同。所以对于异常质量分数采取有损离散化分箱处理,对应的解压缩策略便是该策略的逆过程;
[0048]
步骤五四、采用从头解压缩的策略对整型信息进行解压缩,得到起始比对位置信息start和终止比对位置信息end。
[0049]
整型信息主要是整型位置信息包含序列的比对起始信息,序列的比对终止信息,长度信息等。解压缩过程中采用从头解压缩的策略,解压缩出来的序列按照列占据着每一个位点,在解压缩过程中只需要设置一系列计数器就可以很好地还原出所有位置信息,无需额外的进行存储。由于解压缩过程中会引入参考基因组作为参照,只需要存储整个测序序列中最小起始位置信息,剩余信息在解压缩过程中可以按需求逐一进行再现。通过前后两列长度的差值,表示处理序列数量的变换情况。若第i列长度l
i
小于第i+1列长度l
i+1
,这就表明有l
i+1
‑
l
i
条序列加入到内存中。通过与最小起始位置信息的相对距离,计算得到起始比对位置信息start。如果l
i
>l
i+1
,此时有l
i
‑
l
i+1
条序列完成解压缩,记录终止位置为
‘3’
序列与最小起始位置的相对距离,计算得到终止比对位置end。通过|end
‑
start|可以得到短读序列的长度信息。
[0050]
具体实施方式九:结合图2说明本实施方式。本实施方式是对具体实施方式八的进
一步具体说明,所述步骤五一的具体过程为:
[0051]
对碱基序列进行解压缩,则解压缩比特数据所代表的游程编码数据将组成数据集合{0,1,2,3};
[0052]
字符
‘0’
表示该位点与标准的参考基因组在该位点的碱基一致,为{a,t,c,g}其中之一,该位点是匹配碱基序列;
[0053]
字符
‘1’
表示该位点与标准的参考基因组在该位点的碱基不同,或该位点是未知碱基
‘
n’,通过搜索异常值哈希表在该位点上填补短读序列
‘1’
所对应的异常碱基序列;
[0054]
当字符
‘2’
不连续出现时,则表示该位点存在异常碱基序列,当字符
‘2’
连续出现时,则表示该位点无碱基序列覆盖;
[0055]
字符
‘3’
表示一条完整序列的终止结束。
[0056]
每一个位点的碱基序列依据前一列存储的索引结构,进行dtpbwt解压缩变换,与前一列碱基对齐,形成相对位置测序序列。解压缩过程中,先是通过结尾字符
‘2’
的数量判断该列的匹配碱基序列和异常碱基序列的组成情况。然后通过参考基因组还原
‘0’
所对应的匹配碱基,接下来搜索每一列对应的哈希表,填补上短读序列
‘1’
所对应的异常碱基。当遇到字符
‘3’
时,字符
‘3’
所对应的序列解压完成,去除字符
‘3’
,并将该序列进行存储。最后比对到参考基因组上还原成{a,t,c,g}的字符形式。
[0057]
具体实施方式十:本实施方式是对具体实施方式九的进一步具体说明,所述按照步骤一配置的解压缩输出模式参数进行输出,输出数据的长度为|end
‑
start|。
[0058]
dtpbwt算法区别于tpbwt解压缩算法采用了两大策略,边解压边还原和内置序列
‘
列’索引信息。
[0059]
1)边解压边还原。解压缩过程假设压缩序列文件的覆盖范围是从start
min
到end
max
,所需要显示的区间是start至end,局部解压缩和全局解压缩将压缩文件二进制转换为字符文件需要时间分别为t1,t2,可知t1,t2与解压缩序列长度成正比,且满足以下公式
[0060]
t2=t1(end
‑
start
min
)/end
max
‑
start
min
)
[0061]
局部解压缩在二进制文件还原与全局解压缩上采用相同的算法,所以解压缩时间t
∝
length,当end越小,时间消耗将会越小。序列还原过程dtpbwt算法只需要还原[start,end]之间序列即可。假设tpbwt解压缩和dtpbwt解压缩第二阶段时间分别是t3,t4,那么满足以下公式:
[0062]
t4=t3end
‑
start)/end
max
‑
start
min
)
[0063]
从而推导出总时间t比值公式,当start位置趋向于start
min
时,时间消耗完全与解压缩区间的长度相关。
[0064][0065]
2)内置序列
‘
列’信息
[0066]
每一列结尾存储
‘2’
标识符,并且将每列组成信息压缩到字符
‘2’
的游程编码中。解压缩过程快速定位到所需要区间的时间复杂度是o(n)=o(t(n))=o((end)c),所以还原区间内正常质量分数和异常质量分数的时间复杂度为o(cn)=o(n);还原异常值的时间复杂度为o(cnlogm)=o(n),m=|{a,t,c,g}|=4,c为异常值的长度。对于该部分信息的还原,时间和终止成正比t
∝
end。
[0067]
本发明的上述算例仅为详细地说明本发明的计算模型和计算流程,而并非是对本发明的实施方式的限定。对于所属领域的普通技术人员来说,在上述说明的基础上还可以做出其它不同形式的变化或变动,这里无法对所有的实施方式予以穷举,凡是属于本发明的技术方案所引伸出的显而易见的变化或变动仍处于本发明的保护范围之列。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1