TCO缀合物和治疗剂递送方法与流程
本申请要求2014年11月21日提交的美国临时申请号62/083,022、2014年6月18日提交的62/013,994以及2014年3月14日提交的61/953,294的优先权,各个以全部内容并入文本用于所有目的。
关于对在联邦政府资助的研发下作出的发明的权利的声明
不适用。
技术实现要素:
在第一实施方式中,本发明提供一种固相支持体组合物,具有生物相容的固相支持体,至少一结合(剂),以及具有约1至约10个连接原子的连接体,每个结合剂共价连接至生物相容的固相支持体。
在另一实施方式中,本发明提供一种生物活性组合物,其具有治疗或诊断剂,可以是反式环辛烯或四嗪的结合剂,以及具有约1至约10个连接原子的连接体,结合剂共价连接至治疗或诊断剂。
在另一实施方式中,本发明提供
的组合物。
在另一实施方式中,本发明提供用于选择性递送有效量的治疗剂或诊断剂至患者的目标器官或组织的第一位置的方法。该方法包括在患者的目标器官或组织的第一位置植入本发明的固相支持体组合物,其中,固相支持体组合物包含第一结合剂。该方法还包括给患者施用本发明的生物活性组合物,其中,生物活性组合物包括第二结合剂,且其中第一和第二结合剂在接触后彼此结合,从而选择性递送有效量的治疗或诊断剂至患者的目标器官或组织的第一位置。
在另一实施方式中,本发明提供用于选择性递送有效量的治疗剂或诊断剂至患者的目标器官或组织的第一位置的方法。该方法包括在患者的目标器官或组织的第一位置植入固相支持体组合物,其具有生物相容的固相支持体和连接到固相支持体的第一结合剂。该方法还包括给患者施用生物活性组合物,其具有治疗剂或诊断剂、与第一结合剂互补的第二结合剂以及连接治疗或诊断剂和第二结合剂的可释放的连接体,以使第一和第二结合剂在接触后彼此结合。该方法还包括释放诊断或治疗剂,从而将诊断或治疗剂递送至目标器官或组织的第一位置。
附图简述
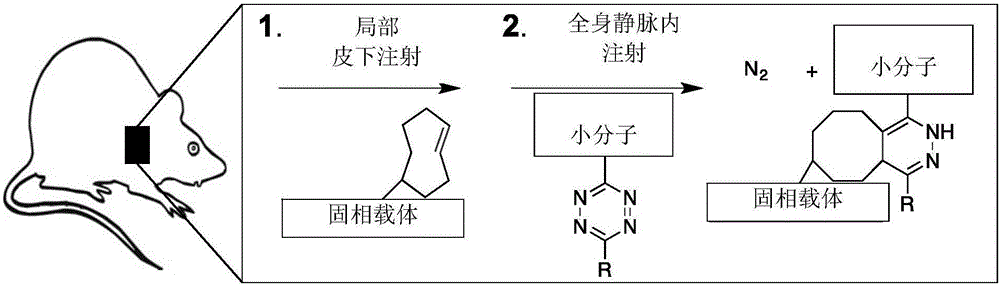
图1A和图1B示出本发明的方法,涉及初始注射共价连接到TCO的生物材料。随后是全身注射偶联到四嗪部分的治疗剂。当两个实体接近时,它们共价反应连接治疗至固相载体,从而定位治疗剂。含有放射性111Im(铟)的Tz放射性探针2用作分子探针。
图2示出体外研究,预称重的凝胶盘(实验或对照)与Tz放射性探针2的溶液混合10分钟或14小时。在10分钟的暴露期,样本TCO-凝胶1结合1.78nmoles/g Tz放射性探针2,而对照凝胶维持0.75nmoles/g Tz放射性探针2。在14小时内,连接到对照凝胶的Tz放射性探针的量保持不变为0.86nmoles/g。TCO-凝胶1结合2.78nmoles/g。误差条代表三个重复实验的平均值的标准偏差。星号表示经配对t检验的统计显著性(p值<0.05)。
图3A和图3B显示Tz放射性探针2的生物分布结果。a、接受皮下TCO-凝胶1(平均0.23克[0.10-0.29克])和对照凝胶(平均0.23克[0.16-0.25克])的小鼠,经尾静脉注射Tz放射性探针2(平均44μCi[39.2–48.4μCi])。b、在特定时间点TCO-凝胶1与对照凝胶的平均差异。柱体表示在特定时间点(1,4,24,48小时)每个器官的%ID/克。误差条表示三个重复实验的平均值的标准偏差。星号表示经配对t检验的统计显著性(p值<0.05)。
图4示出接受皮下TCO-凝胶1(170mg,绿色区域)和对照凝胶(230mg,黄色区域)的小鼠。植入后3小时,经尾静脉注射递送Tz放射性探针2(1.05mCi)并通过SPECT在4,24,48小时成像。
图5示出固相支持体组合物TZ-Me-凝胶的合成制备。
图6示出通过非可释放的连接体与TCO连接的荧光素与四嗪固相支持体之间的反应。R代表完整二乙胺荧光素部分,仍共价地连接到固相支持体。
图7示出通过可释放的连接体与TCO连接的荧光素与四嗪固相支持体之间的反应。如所预期的,荧光素二乙胺(MW:418.40)和二氧化碳从固相支持体释放。
图8提供除去凝胶后的上清液样品,用于推断未反应的TCO-X-荧光素的量及TCO-X-荧光素和TZ-Me-凝胶之间的反应动力学。简言之,方法涉及:形成和称重Tz-凝胶,加入1毫升含预定浓度的TCO-X-荧光素(10,20,50纳摩尔)的PBS盐水,将板置于孔板混合器(速度200)预定量的时间(10,50,100,180分钟),在特定时间点转移上清液至另一板,和通过IVIS光谱仪检测残留在上清液中荧光的量(490nm激发荧光素,515nm发射)。
图9示出基于实验值的荧光团量和相关的辐射(荧光)的标准曲线拟合,其中利用已知纳摩尔量的TCO-NR-荧光素,用IVIS光谱仪检测的辐射(490nm激发荧光素,515nm发射)。
图10示出当和TZ-Me-凝胶在37℃混合0至70分钟时,上清液中TCO-NR-荧光素随时间减少。
图11示出结合到凝胶的TCO-NR-荧光素和上清液中检测到的未结合的量的理论相互性。
图12示出根据图10的数据,结合到TZ-Me-凝胶的TCO-NR-荧光素分子的计算量。基于给出的数据,当TZ-Me-凝胶盘与50纳摩尔TCO-NR-荧光素混合时,70分钟后,每1毫克TZ-Me-凝胶有0.66nmolesTCO-NR-荧光素反应(0.66nmoles/mg)。即使只在70分钟后,是如图2所示用TCO-凝胶14小时后获得的量(2.78nmoles/g)的约200倍。
图13显示在37℃孵育预定量时间(0-3天)后TZ-Me-凝胶的活性。在规定的孵育后,加入50纳摩尔TCO-NR-荧光素激发50毫克凝胶90分钟,依照前图计算结合量。
图14示出将TCO-R-荧光素加到TZ-Me-凝胶多个时间点后未结合分子的预期与观察量。考虑到TCO-R-荧光素和TCO-NR荧光素之间的相似之处,预计反应模式类似。假设是上清液中回收荧光的量的差异是由于二乙基荧光素的释放,如图7所示。
图15示出将特定量TCO-R-荧光素(10、20、50纳摩尔)加入TZ-Me-凝胶后释放的荧光素的总量。此外,采用LC-MS分析在时间点60分钟上清液中化合物的具体特性并显示存在荧光素二胺(分子量418.40)为主要化合物,还证实存在TCO-R-荧光素与TZ-Me-凝胶的捕获和释放反应的消除产物。
图16A和图16B示出使用捕获和释放连接体的本发明的方法。简要地说,该方法首先包括含点击化学载体改性的固体载体的定位,在该特定实例中为四嗪。接着氨基甲酸酯(绿色)和反式环辛烯基团改性的载物暴露于材料(无论体外或体内)。两试剂原位反应,根据四嗪取代基,载物或从固体载体释放或固定到固体载体。
图17示出暴露于不同的四嗪修饰的琼脂糖珠后TCO-R-罗丹明的释放动力学。简言之,如图16所示在X和Y具有杂环取代基的琼脂糖1a,导致缓慢释放(红线),大概是由于吸电子基团引起的衰减。另一方面,在X上具有芳族取代基和在Y上具有烷基取代基导致罗丹明二乙胺的快速释放。值是三次实验的结果。
图18示出未植入凝胶或植入Tz-凝胶,或仅植入海藻酸,然后注入TCO-R-罗丹明或TCO-NR-罗丹明的小鼠在70分钟,5小时和3天辐射值。
图19A和图19B示出TCO-R-万古霉素对发光MSSA的MIC(Xen 29)。
图20示出(R,E)-N-(2-氨乙基)-2-(环辛-4-烯-1-基氧)乙酰胺(5)的NMR。
图21示出未改性海藻酸凝胶的NMR。
图22示出TCO-凝胶1的NMR。
图23示出Tz-Me-凝胶的NMR。
图24示出“捕获和释放”策略,采用生物正交TCO-四嗪化学。
图25示出180分钟后荧光素释放量。计算为结合荧光团的预期值和获得的实际实验值之间的差。
图26示出60分钟后Tz-Me-凝胶和TCO-R-荧光素混合物上清液中发现的荧光素二胺的峰([M+H]419.1214和[M+MeOH]453.3417),通过液相色谱-质谱确定预期的“捕获和释放”产品。
图27示出暴露于不同四嗪改性的琼脂糖珠后TCO-R-荧光素([M+H]is 571.21)的释放动力学。简言之,如图16所示在X和Y具有杂环取代基的琼脂糖1a,导致缓慢释放(MX53,红线),大概是由于吸电子基团引起的衰减。另一方面,在X上具有芳族取代基和在Y上具有烷基取代基导致罗丹明二乙胺(MX65,蓝线)的快速释放。左手边示出通过质谱动态评估释放。
图28示出荧光素标记的TCO从四嗪改性琼脂糖的“捕获和释放”的动力学研究设计。
图29示出通过质谱学方法动态评估TCO-R-罗丹明([M+H]625.31)的释放。如所预期在473.22发现释放产物。
图30示出基于以前的动力学生物分布研究,注射TCO-R-罗丹明后在各时间点比较阴性对照(无凝胶注射)和释放方案(TZ-Me-凝胶)荧光的增加。柱状图推断8小时内获得推定总量的数据。在凝胶存在下的曲线下面积反映的剂量几乎是双倍的,如“释放”柱(右)相对阴性对照(左)所示。
图31示出小鼠的动力学多剂量评估。在各自通过尾静脉注射50纳摩尔TCO-R-罗丹明后对三只小鼠进行评价。示出如上所述处理小鼠的图像。“阴性对照”对象不接受任何凝胶注射。“释放”对象在背部皮下接受Tz-Me-凝胶注射。“凝胶对照”对象在背部皮下接受海藻酸凝胶注射。在每次给荧光团后5-10分钟和110-150分钟后用IVIS光谱仪评价小鼠。
图32示出在给每个不同实验组(阴性对照=无凝胶;实验组=Tz-Me-凝胶;凝胶对照=未改性海藻酸凝胶)第二、第三和第四次注射50纳摩尔TCO-R-罗丹明后5-10分钟相较于110-150分钟所观察到的平均荧光值。实验组荧光在统计学上高于阴性或凝胶对照组。通过配对t检验进行比较(n=3)。
具体实施方式
I.概述
本发明提供用于植入和给药于对象的组合物,其中组合物使用短连接体,其也可以包括可释放的组分。可释放的连接体能将更大量的治疗剂或诊断剂给药至有需要的患者。
II.定义
“治疗剂”是指能够治疗和/或减轻病症或疾病的试剂。代表性的治疗剂包括但不限于,紫杉醇,多柔比星,依托泊苷,伊立替康,SN-38,环孢菌素A,鬼臼毒素,卡氮芥,两性霉素,伊沙匹隆,帕妥匹隆(埃博霉素epothelone类),万古霉素,雷帕霉素和铂类药物。本发明的治疗剂还包括前药形式。
“诊断剂”是指帮助诊断病症或疾病的试剂。代表性的诊断剂包括成像剂,如顺磁性试剂,光学探针和放射性核素。顺磁性试剂是在外加场下具有磁性的成像剂。顺磁性试剂的例子包括但不限于,铁颗粒包括纳米颗粒。光学探针是可以通过一辐射波长激发并在第二、不同的辐射波长检测进行检测的荧光化合物。本发明中有用的光学探针包括但不限于,Cy5.5、Alexa(亚历克萨)680、Cy5、DiD(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚二碳菁高氯酸盐)和DiR(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚三碳菁碘化物)。其他光学探针包括量子点。放射性核素是经过放射性衰变的元素。本发明中有用的放射性核素包括但不限于:3H、11C、13N、18F、19F、60Co、64Cu、67Cu、68Ga、82Rb、90Sr、90Y、99Tc、99mTc、111In、123I、124I、125I、129I、131I、137Cs、177Lu、186Re、188Re、211At、Rn、Ra、Th、U、Pu和241Am。
“目标器官或组织”是指作为目标用于递送治疗或诊断剂的器官或组织。作为目标的代表性的器官和组织包括能被化学或生物靶向剂所靶向的那些,以及不能被化学或生物靶向剂靶向的那些器官和组织。代表性的器官或组织包括骨。
“选择性递送”是指递送治疗或诊断剂至需要治疗的器官或组织部分,而不靶向不需要治疗的器官或组织的其他部分。
“移植”是指手术植入到患者体内。
“生物相容的固相支持体”是指能够植入患者体内且支持结合剂之一,以及结合剂结合后的治疗或诊断剂的固相支持体材料。固相支持体与患者身体相容。代表性的生物相容的固相支持体包括但不限于,水凝胶如多糖水凝胶,海藻酸,纤维素,壳聚糖,透明质酸,硫酸软骨素,肝素等。
“接触的”或“接触”是指使两不同物种接触以使它们能反应的过程。但应理解,得到的反应产物可以直接产生自添加试剂间的反应或产生自能够在反应混合物中产生的一个或更多添加试剂的中间体。
“连接体”、“连接的”或“连接”是指连接本发明化合物至靶向特定类型细胞,如癌细胞,其他类型疾病细胞或正常细胞类型的生物材料的化学部分。连接可以通过共价或离子键形成。连接可以是两被连接部分直接连接或间接连接,如通过连接体。本发明中有用的连接体长度至多30个碳原子。优选地,连接体长度是5-15个碳原子。用于将接头连接到本发明的化合物和生物分子的键的类型包括但不限于酰胺,胺,酯,氨基甲酸酯,脲,硫醚,硫代氨基甲酸酯,硫代碳酸盐和硫脲。本领域技术人员将认识到其它类型的键在本发明中是有用的。
“结合剂”是指能在生物环境中与另一结合剂形成共价键的任何群组。这通常被称为生物结合或生物正交化学。代表性的结合剂包括但不限于,胺和活化酯,胺和异氰酸酯,胺和异硫氰酸酯,用于形成二硫化物的硫醇,用于形成烯胺的醛和胺,用于通过施陶丁格连接形成酰胺的叠氮化物,用于经由点击-化学形成三唑的炔烃和叠氮化物,反式-环辛烯(TCO)和四嗪等等。本发明中有用的结合剂与相应的结合剂具有高反应性,以致迅速反应。
“治疗”,“治疗的”和“治疗方法”是指治疗或改善损伤,病理,病症或症状(例如,疼痛)的任何成功的标记,包括任何客观或主观的参数,如症状的减轻、缓解、减弱或使得患者更可容忍症状、损伤、病理或病症;降低病症或症状频率或持续时间;或者,在某些情况下,预防症状或病症的发作。症状的治疗或改善可以基于任何客观或主观参数,包括例如身体检查的结果。
“给药”是指口服给药,以栓剂给药,局部接触,胃肠外,静脉内,腹膜内,肌内,病灶内,鼻内或皮下给药,鞘内给药,或植入缓释装置,如,微型渗透泵,给对象。
“患者”是指需要治疗的动物,如哺乳动物,包括但不限于,灵长类(例如人),牛,绵羊,山羊,马,狗,猫,兔,大鼠,小鼠等。在某些实施方式中,患者是人。
“治疗有效量或剂量”或“治疗足够量或剂量”或“有效或足够量或剂量”是指对被给药者产生治疗效果的剂量。确切的剂量取决于治疗目的,由本领域技术人员使用已知的技术(参见,如Lieberman,药物剂型(Pharmaceutical Dosage Forms)(卷1-3,1992);Lloyd,制药工艺,科学与技术(The Art,Science and Technology of Pharmaceutical Compounding)(1999);Pickar,剂量计算(Dosage Calculations)(1999);和Remington:药剂学科学和实践(The Science and Practice of Pharmacy),第20版本,2003,Gennaro编.,Lippincott,Williams和Wilkins)确定。在致敏细胞中,治疗有效剂量通常可以低于非致敏细胞传统的治疗有效剂量。
III.组合物
本发明提供组合物,具有短连接体或可释放的连接体用于选择性递送治疗或诊断剂给患者。在一些实施方式中,本发明提供固相支持体组合物,具有生物相容的固相支持体,至少一结合剂,和具有约1-约10个连接原子的连接体,将各结合剂共价连接至生物相容的固相支持体。
任何合适的生物相容的固相支持体可以用于本发明的方法。例如,生物相容的固相支持体可以是水凝胶、交联聚合物基质、金属、陶瓷、塑料等等。用于本发明的水凝胶包括但不限于,多糖凝胶,海藻酸,纤维素,透明质酸,壳聚糖,甲壳素,几丁质,透明质酸,硫酸软骨素,肝素等。其他糖基生物材料是现有技术已知的,如聚合物先进技术(Polymer Advanced Technology)2014,25,448-460描述的那些。用作生物相容的支持体的聚合物可以包括但不限于:聚磷腈,聚酐,聚缩醛,聚(原酸酯),聚磷酸酯,聚己内酯,聚氨酯,聚乳酸,聚碳酸酯,聚酰胺和聚醚,以及它们的共混物/复合材料/共聚合物。代表性的聚醚包括,但不限于,聚(乙二醇)(PEG),聚丙二醇(PPG),三嵌段聚醚(Pluronic,[PEG]n-[PPG]m-[PEG]n),PEG二丙烯酸酯(PEGDA)和PEG二甲基丙烯酸酯(PEGDMA)。生物相容的固相支持体也可以包括蛋白质和其他聚(氨基酸),例如胶原,明胶,弹性蛋白和弹性蛋白样多肽,白蛋白,纤维蛋白,聚(γ-谷氨酸),聚(L-赖氨酸),聚(L-谷氨酸),和聚(天冬氨酸)。
在一些实施方式中,固相支持体可以是水凝胶。在一些实施方式中,固相支持体可以是海藻酸(盐)。在一些实施方式中,固相支持体可以是几丁质。在一些实施方式中,固相支持体可以是透明质酸。在一些实施方式中,固相支持体可以是壳聚糖。在一些实施方式中,固相支持体可以是琼脂糖。
任何合适的连接体可以用于本发明以连接结合剂至生物相容的固相支持体或治疗或诊断剂。代表性的连接体可以具有约1-约100个连接原子,且可包括乙烯氧基部分,胺,酯,酰胺,酮,脲,氨基甲酸酯和碳酸酯官能团。用于本发明方法的其他连接体可以具有约1-约50个连接原子,或约1-约10个连接原子,或约5-约10个连接原子。代表性的连接体包括但不限于以下示出的那些:
适用于本发明的其他连接体包括:
任何合适的结合剂可以用于本发明的方法。可以在生物偶联技术“Bioconjugate Techniques”Greg T.Hermanson,1996和ACS化学生物学(ACS Chemical Biology)2014,9,592-605中找到代表性的结合剂。例如,用于本发明方法的结合剂包括但不限于,环辛烯,四嗪,叠氮化物,炔,胺,活化的酯,异氰酸酯,异硫氰酸酯,硫醇,醛,酰胺,等等。在一些实施方式中,结合剂可以是环辛烯,四嗪,叠氮化物或炔。在一些实施方式中,结合剂可以是反式-环辛烯或1,2,4,5-四嗪。在一些实施方式中,结合剂可以是反式-环辛烯。在一些实施方式中,结合剂可以是4-甲基-1,2,4,5-四嗪。
在一些实施方式中,结合剂和连接体共同结构为:
在一些实施方式中,组合物结构为:
本发明还提供通过连接体连接到结合剂的治疗剂或诊断剂的组合物。在一些实施方式中,本发明提供生物活性组合物,具有治疗或诊断剂、可以是反式-环辛烯或四嗪的结合剂,以及具有约1-约10个连接原子的连接体,共价连接结合剂至治疗或诊断剂。
任何治疗或诊断剂可以用于本发明的方法。代表性的治疗剂包括但不限于,抗生素如万古霉素,紫杉醇,多柔比星,依托泊苷,伊立替康,SN-38,环孢菌素A,鬼臼毒素,卡氮芥,两性霉素,伊沙匹隆,帕妥匹隆(埃博霉素epothelone类),雷帕霉素和铂类药物。其它治疗剂包括多西环素和其它MMP抑制剂。仍然其它治疗剂包括达托霉素,左旋多巴,奥司他韦,头孢氨苄,5-氨基乙酰丙酸,半胱氨酸,制霉菌素,两性霉素B,氟胞嘧啶,恩曲他滨,甲氧苄啶,磺胺甲恶唑,阿昔洛韦,塞来考昔,尼莫地平,强力霉素,头孢曲松,等等。在一些实施方式中,治疗剂可以是万古霉素。在其他实施方式中,治疗剂可以是达托霉素。在另外的实施方式中,治疗剂可以是阿霉素。在另一实施方式中,治疗剂可以是环磷酸腺苷(c-AMP)。
代表性的诊断剂包括成像剂,如顺磁性试剂,光学探针和放射性核素。顺磁性试剂在外加场下具有磁性的成像剂。顺磁性试剂的例子包括但不限于,铁颗粒包括纳米颗粒。光学探针是可以通过一辐射波长激发并在第二、不同的辐射波长检测进行检测的荧光化合物。本发明中有用的光学探针包括但不限于,Cy5.5、Alexa(亚历克萨)680、Cy5、DiD(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚二碳菁高氯酸盐)和DiR(1,1'-二(十八烷基)-3,3,3',3'-四甲基吲哚三碳菁碘化物)。其他光学探针包括量子点。放射性核素是经过放射性衰变的元素。本发明中有用的放射性核素包括但不限于:3H、11C、13N、18F、19F、60Co、64Cu、67Cu、68Ga、82Rb、90Sr、90Y、99Tc、99mTc、111In、123I、124I、125I、129I、131I、137Cs、177Lu、186Re、188Re、211At、Rn、Ra、Th、U、Pu和241Am。诊断剂也可以包括螯合剂,如1,4,8,11-四氮杂环十二烷-1,4,8,11-四乙酸(TETA)、4,11-双(羧甲基)-1,4,8,11-四氮杂双环[6.6.2]十六烷(CB-TE2A)、二乙烯三胺五乙酸(DTPA)和1,4,7,10-四-氮杂环癸烷四乙酸(DOTA)。其它螯合剂在本发明的方法中是有用的。
任何合适的连接体可以用于本发明从而将结合剂连接到生物相容的固相支持体或治疗或诊断剂。代表性的连接体可以具有约5到约100个连接原子,且可以包括乙烯-氧部分,胺,酯,酰胺,酮,脲,氨基甲酸酯和碳酸酯官能团。本发明的方法中有用的其它连接体可具有约5至约50个连接原子,或约1至约10个连接原子,或约5至约10个连接原子。代表性的连接体包括但不限于,以下示出的那些:
任何合适的结合剂可以用于本发明的方法。可以在生物偶联技术“Bioconjugate Techniques”Greg T.Hermanson,1996和ACS化学生物学(ACS Chemical Biology)2014,9,592-605中找到代表性的结合剂。例如,用于本发明方法的结合剂包括但不限于,环辛烯,四嗪,叠氮化物,炔,胺,活化的酯,异氰酸酯,异硫氰酸酯,硫醇,醛,酰胺,等等。在一些实施方式中,结合剂可以是环辛烯,四嗪,叠氮化物或炔。在一些实施方式中,结合剂可以是反式-环辛烯或1,2,4,5-四嗪。在一些实施方式中,结合剂可以是反式-环辛烯。在一些实施方式中,结合剂可以是4-甲基-1,2,4,5-四嗪。
在一些实施方式中,结合剂和连接体共同结构为:
在一些实施方式中,本发明提供的组合物为
IV.选择性递送治疗或诊断剂的方法
本发明提供通过将本发明的固相支持体组合物植入有此需要的患者,再将本发明的生物活性组合物给药于患者的选择性递送治疗或诊断剂的方法。在一些实施方式中,本发明提供选择性递送有效量的治疗或诊断剂到患者目标器官或组织的第一位置的方法。方法包括将本发明的固相支持体组合物植入患者目标器官或组织的第一位置,其中固相支持体组合物包括第一结合剂。方法还包括将本发明的生物活性组合物给药于患者,其中,所述生物活性组合物包括第二结合剂,其中第一和第二结合剂在接触后互相结合,从而选择性递送有效量的治疗或诊断剂到患者目标器官或组织的第一位置。
使用本发明的方法可以靶向任何合适的器官或组织。代表性的器官或组织包括但不限于,骨,软骨,韧带,肌腱,肠,肌肉,神经系统包括脑,脊髓,心脏,神经和其他。例如,当器官是心脏,本发明的方法可用于心脏修复。在一些实施方式中,患者目标器官或组织的第一位置相对于目标器官或组织的其他位置不能有选择地通过化学或生物靶向剂打靶。在一些实施方式中,目标器官或组织可以是骨。
在以上为固相支持体组合物和生物活性组合物的更多详述中描述了适于本发明方法的结合剂。在一些实施方式中,第一结合剂可以是反式-环辛烯,第二结合剂可以是四嗪。在一些实施方式中,第二结合剂可以是1,2,4,5-四嗪。在一些实施方式中,第二结合剂可以是4-甲基-1,2,4,5-四嗪。
在一些实施方式中,可植入的组合物具有结构:
在一些实施方式中,可植入的组合物可以是:
和
生物活性组合物可以是:
其中R可以是治疗剂或诊断剂。
可以通过本领域技术人员已知的任何方式植入生物相容的固相支持体。
治疗或诊断剂可以足以治疗患者患有的疾病或病症的任何合适的量给药。给药的剂量、频率和时间将很大程度上取决于所选择的治疗剂,待治疗的病症的性质,患者的情况包括年龄、体重和其他病症或疾病症的存在,待给药的制剂和主治医师的判断。通常,治疗剂或诊断剂以每天约2毫克到至多约2000毫克的剂量给药,然而必然将发生如上所述取决于疾病靶,患者和给药途径的变化。本发明的治疗或诊断剂可以需要的频率给药,包括每小时,每天,每周或每月。本发明的制剂方法中使用的治疗或诊断剂以每天约0.0001mg/kg到约1000mg/kg的初始剂量给药。可以使用每日剂量范围约0.01mg/kg到约500mg/kg,或约0.1mg/kg到约200mg/kg,或约1mg/kg到约100mg/kg,或约10mg/kg到约50mg/kg。但是,剂量可以根据患者的要求、治疗病症的严重性和使用的化合物而变化。例如,考虑特定患者诊断疾病的类型和阶段凭经验确定剂量。施用给患者的剂量,在本发明的上下文中,应随时间足以产生有益的治疗效果,但通常低于没植入将治疗或诊断剂聚集在需要治疗器官或组织处的生物相容的固相支持体情况下治疗患者所需的剂量。剂量大小还将由伴随将特定化合物给药于特定患者的任何不良副作用的存在,性质和程度来确定。特定情况的合适剂量的确定在医师的技术范围内。通常,治疗开始时用较小的剂量,它小于化合物的最佳剂量。此后,以小的增量增加剂量直至达到情况下的最佳效果。为方便起见,如果需要,总日剂量可以在一天分次部分给药。剂量可每日给予,或隔天,如由治疗医生所确定的。也可以经更长的时间(周,月或年)定期或连续给予剂量。
在一些实施方式中,目标器官或组织的第一位置的治疗或诊断剂的浓度高于患者其他位置的浓度。在一些实施方式中,治疗剂是万古霉素。
在另一实施方式中,本发明提供选择性递送有效量的治疗或诊断剂到患者目标器官或组织的第一位置的方法。方法包括在患者目标器官或组织的第一位置植入具有生物相容的固相支持体和连接到所述固相支持体的第一结合剂的固相支持体组合物。方法还包括将具有治疗或诊断剂,与第一结合剂互补的第二结合剂以及连接治疗或诊断剂和第二结合剂的可释放的连接体的生物活性组合物给药至患者,以致第一和第二结合剂在接触后互相结合。方法还包括释放治疗或诊断剂,从而将治疗或诊断剂递送至目标器官或组织的第一位置。
V.制剂
本发明的组合物可以以各种各样的口服,胃肠外和局部剂型来制备。口服制剂包括适合于患者咽下的片剂,丸剂,粉末,糖衣丸,胶囊,液体,锭剂,扁囊剂,凝胶,糖浆剂,膏剂,混悬剂等。本发明的组合物也可以通过注射,即通过静脉,肌内,皮内,皮下,十二指肠内,或腹腔内给药。此外,本文描述的组合物可通过吸入给药,例如,鼻内给药。另外,本发明的组合物可以经皮给药。本发明的组合物还可以通过眼内,阴道内,直肠内给药,包括栓剂、吹入剂、粉末和气雾剂(类固醇吸入剂的例子,见Rohatagi,临床药理学杂志“J.Clin.Pharmacol.”35:1187-1193,1995;Tjwa,过敏哮喘免疫学年报“Ann.Allergy Asthma Immunol.”75:107-111,1995)。此外,本发明还提供药物组合物,包括药学上可接受的载体或赋形剂以及本发明的化合物。
为从本发明化合物制备药物组合物,药学上可接受的载体可以是固体或液体。固体形式制剂包括粉剂,片剂,丸剂,胶囊剂,扁囊剂,栓剂和可分散颗粒剂。固体载体可以是一种或多种物质,其也可以作为稀释剂,调味剂,粘合剂,防腐剂,片剂崩解剂,或包封材料。关于制剂和给药技术的细节在科学和专利文献中有很好的描述,见,例如,雷明顿Remington的药物科学的最新版本,Maack出版公司,伊斯顿PA(“雷明顿的”)。
在粉剂中,载体是细碎的固体,其在含细碎活性组分的混合物中。在片剂中,活性组分以合适的比例与具有所需性能的载体混合并被压实成所需的形状和大小。粉剂和片剂优选含有5%或10%到70%的本发明化合物。
合适的固体赋形剂包括但不限于,碳酸镁;硬脂酸镁;滑石;果胶;糊精;淀粉;黄蓍胶;低熔点蜡;可可脂;碳水化合物;糖,包括但不限于,乳糖,蔗糖,甘露醇或山梨糖醇;来自玉米,小麦,水稻,马铃薯或其他植物的淀粉;纤维素,如甲基纤维素,羟丙基甲基纤维素,或羧甲基纤维素钠;和树胶,包括阿拉伯和黄蓍胶;以及蛋白,包括但不限于,明胶和胶原蛋白。如果需要,可加入崩解剂或增溶剂,如交联聚乙烯吡咯烷酮,琼脂,海藻酸或其盐,如海藻酸钠。
糖衣丸芯具有合适的包衣如浓缩的糖溶液,其还可以含有阿拉伯胶,滑石,聚乙烯吡咯烷酮,卡波姆凝胶,聚乙二醇和/或二氧化钛,漆液和合适的有机溶剂或溶剂混合物。染料或色素可加入片剂或糖衣丸包衣用于产品识别或表征活性化合物的量(即,剂量)。也可以口服使用本发明的药物制剂,例如使用如甘油或山梨糖醇的包衣和明胶制成的软的,密封胶囊,以及明胶制成的推入配合胶囊。推入配合胶囊可以含有本发明的化合物,混合有填充剂或粘合剂如乳糖或淀粉,润滑剂如滑石粉或硬脂酸镁,和任选的稳定剂。在软胶囊中,本发明的化合物可以溶解或悬浮在合适的液体中,例如脂肪油,液体石蜡,或液体聚乙二醇,有或没有稳定剂。
为了制备栓剂,首先将低熔点蜡,如脂肪酸甘油酯或可可脂的混合物融化,本发明的化合物均匀地分散其中,例如通过搅拌。然后将熔化的均匀混合物倒入适宜大小的模具中,使其冷却并由此固化。
液体形式制剂包括溶液,悬浮液和乳液,例如,水或水/丙二醇溶液。对于肠胃外注射,液体制剂可以在水性聚乙二醇溶液中配制成溶液。
适合于口服使用的水溶液可通过将本发明的化合物溶解在水中并根据需要加入合适的着色剂,调味剂,稳定剂和增稠剂来制备。适于口服使用的水性混悬液可以通过将细碎的活性组分分散在水中制成,水中含有粘性物质如天然或合成树胶,树脂,甲基纤维素,羧甲基纤维素钠,羟丙基甲基纤维素,海藻酸钠,聚乙烯吡咯烷酮,黄蓍树胶和阿拉伯树胶,和分散或润湿剂如天然存在的磷脂(例如,卵磷脂),环氧烷烃与脂肪酸的缩合产物(例如,聚乙二醇硬脂酸酯),环氧乙烷与长链脂肪醇的缩合产物(例如,十七乙烯氧基十六醇),环氧乙烷与衍生自脂肪酸和己糖醇的偏酯的缩合产物(如,聚氧乙烯山梨糖醇单油酸酯),或环氧乙烷与衍生自脂肪酸和己糖醇酐的偏酯的缩合产物(如,聚氧乙烯山梨糖醇单油酸酯)。水性悬浮液也可含有一种或多种防腐剂例如对羟基苯甲酸乙酯或正丙酯,一种或多种着色剂,一种或多种调味剂和一种或多种甜味剂,如蔗糖,阿司帕坦或糖精。制剂可用渗透度来调节。
还包括固体形式制剂,其在使用之前不久预期转换为液体形式制剂用于口服给药。这样的液体形式包括溶液,悬浮液和乳液。除了活性成分外,这些制剂可以含有着色剂,调味剂,稳定剂,缓冲剂,人工和天然甜味剂,分散剂,增稠剂,增溶剂等。
在另一实施方式中,可以配制本发明的组合物用于肠胃外给药,如静脉内(IV)施用或给药到器官的体腔或内腔。用于给药的制剂通常包括本发明的组合物溶解于药学上可接受的载体形成的溶液。能够被使用的可接受的载体和溶剂有水和林格氏溶液,等渗氯化钠。此外,无菌固定油可以常规地用作溶剂或悬浮介质。为了这个目的,可以采用任何温和的固定油,包括合成的单或二甘油酯。此外,脂肪酸如油酸同样可用于注射剂的制备中。这些溶液是无菌的,并且通常没有不期望的物质。这些制剂可以通过常规的,众所周知的灭菌技术进行灭菌。当需要接近生理条件时制剂可含有药学上可接受的辅助物质,例如pH调节剂和缓冲剂,毒性调节剂,例如,乙酸钠,氯化钠,氯化钾,氯化钙,乳酸钠等。本发明的组合物在这些制剂中的浓度可以广泛变化,并且将根据选择的特定给药模式和患者的需求主要基于流体体积,粘度,体重等进行选择。对于IV给药,制剂可以是无菌可注射的制剂,如无菌可注射水性或油性悬浮液。这种悬浮液可以根据已知技术用那些适当的分散剂或湿润剂和悬浮剂配制。无菌可注射制剂还可以是在无毒的胃肠外可接受的稀释剂或溶剂中的无菌注射溶液或悬浮液,例如1,3-丁二醇溶液。
在另一实施方式中,本发明的组合物的制剂可以通过使用与细胞膜融合或被内吞的脂质体递送,即通过采用附着到脂质体或直接附着到寡核苷酸的配体,其结合细胞的表面膜蛋白受体导致内吞。通过使用脂质体,特别是在脂质体表面带有特异于靶细胞的配体的位置,或以其它方式优先定向特定器官,可以将本发明的组合物集中递送到体内的靶细胞(参见例如,Al-Muhammed,微胶囊杂志“J.Microencapsul.”13:293-306,1996;Chonn,Curr.Opin.Biotechnol.6:698-708,1995;Ostro,Am.J.Hosp.Pharm.46:1576-1587,1989)。
VI.给药
本发明的组合物可以通过任何合适的方式递送,包括口服,肠道外和局部方法。经皮给药方法,通过局部途径,可以配制成涂药棒,溶液,悬浮液,乳液,凝胶,霜剂,软膏剂,糊剂,胶冻剂,涂料,粉末和气雾剂。
药物制剂优选为单位剂型。在这样的形式中,制剂被细分为含有合适量本发明化合物的的单位剂量。单位剂型可以是包装的制剂,含有离散量制剂的包装,如包装的片剂,胶囊和在小瓶或安瓿中的粉剂。此外,单位剂型可以是胶囊,片剂,扁囊剂或锭剂本身,或者它可以是合适数量的这些的包装形式。
本发明的化合物可以任何合适的量存在,并且可以取决于各种因素,包括但不限于,对象的体重和年龄,疾病的状态等。本发明化合物的合适剂量范围包括约0.1毫克至约10,000毫克,或约1mg至约1000mg,或约10毫克到约750毫克,或约25毫克到约500毫克,或约50毫克至约250毫克。本发明的化合物的合适剂量包括约1毫克、5、10、20、30、40、50、60、70、80、90、100、200、300、400、500、600、700、800、900或1000毫克。
本发明的化合物可以任何合适的频率,间隔和持续时间给药。例如,本发明的化合物可以一次一小时给药,或一个小时两次、三次或更多次,每天一次,或每天两次、三次或更多次,或每2,3,4,5,6或7天一次,以便提供优选的剂量水平。当本发明的化合物给药多于每日一次,代表性的间隔包括5,10,15,20,30,45和60分钟,以及1,2,4,6,8,10,12,16,20和24小时。本发明的化合物可以是施用一次,两次,或三次或更多次,给药一小时,1-6小时,1至12小时,1至24小时,6至12小时,12至24小时,一天,1至7天,单周,1至4周,一个月,1至12个月,一年或更多,或者甚至无限期。
本发明的化合物可与另一活性剂共同给药。共同给药包括在0.5、1、2、4、6、8、10、12、16、20或24小时彼此施用本发明的化合物和活性剂。共同给药还包括同时,大致同时(例如,彼此在约1、5、10、15、20或30分钟内),或以任何次序顺序地给药。此外,本发明的化合物和活性剂可以各自每天一次,或每天两次、三次或更多次给药,以提供每天优选剂量水平。
在一些实施方式中,共同给药可以通过共制剂实现,即制备包括本发明的化合物和活性剂两者的单一的药物组合物。在其他实施方式中,本发明的化合物和活性剂可以单独配制。
本发明的化合物和活性剂可以任何合适的重量比存在于本发明的组合物中,例如从约1:100至约100:1(重量/重量),或约1:50到约50:1,或约1:25至约25:1,或约1:10至约10:1,或约1:5至约5:1(重量/重量)。本发明的化合物和其他活性剂可以任何合适的重量比存在,例如约1:100(重量/重量)、1:50、1:25、1:10、1:5、1:4、1:3、1:2、1:1、2:1、3:1、4:1、5:1、10:1、25:1、50:1或100:1(重量/重量)。本发明的化合物和活性剂的其他剂量和剂量比在本发明的组合物和方法中适用。
VII.实施例
材料除非另有说明,全部试剂和NMR溶剂购自西格玛-奥尔德里奇Sigma-Aldrich(圣路易斯,密苏里州)。化合物2获自虹膜生物技术Iris Biotech(马克特雷德维茨,德国),而化合物8购自Polypure(奥斯陆,挪威)。DOTA-NHS酯获自Macrocyclics(达拉斯,德克萨斯州)。硅胶购自Silicycle(魁北克,加拿大),而制备TLC板(20x 20cm;厚度1000μm)购自安莱尔科技Analtech(纽瓦克,DE)。超纯海藻酸购自ProNova生物医学(挪威)。[111In]氯化铟溶液购自珀金埃尔默PerkinElmer(沃尔瑟姆,美国)。稀盐酸中的[64Cu]氯化铜购自华盛顿大学(圣路易斯,密苏里州)或采用11MeV西门子RDS 111回旋加速器通过64Ni(p,n)64Cu核反应内部制作并通过阴离子交换色谱法(Biorad AG 1-X8)纯化。Dulbecco杜氏磷酸缓冲盐(DPBS)购自英杰公司(卡尔斯巴德,加利福尼亚最高)。
方法采用瓦里安400MHz VNMRS仪器在CDCL3或[D6]DMSO中进行NMR实验。在波士顿大学化学仪器中心使用安捷伦离子阱LC/MSD SL得到高分辨率ESI质谱数据,无论是阳性或阴性测定。在有机合成阶段,使用装有沃特斯(Waters)XBridge C18柱(19×250毫米)的安捷伦Agilent 1100系列,应用梯度含0.1%TFA的MeCN和水,进行HPLC纯化。
放射化学期间,对于体内分析和纯化,使用装有Jupiter Proteo C-12柱(250×4.6mm,4μm,菲罗门Phenomenex,托伦斯,加利福尼亚州)以及串联至Bioscan公司的FlowCount光电倍增管(PMT)(Bioscan,华盛顿,哥伦比亚特区)的单波长或二极管阵列UV检测器(设置220和254nm)的贝克曼-柯尔特系统金(Beckman-Colter System Gold)128色谱系统实施反相HPLC。使用32Karat软件包(Beckman-Colter)分析数据。流动相由溶剂A:水中0.05%三氟乙酸和溶剂B:100%乙腈构成,1.5毫升/分钟的流速,注射后2分钟开始线性梯度,在30分钟内从9%溶剂B,然后增加至81%,除非另有说明。
使用海藻酸凝胶的体外实验期间,在装有沃特斯2487双吸光度检测器(220和320毫微米)和Bioscan公司的Flow-count放射性检测器的沃特斯微风(Waters Breeze)色谱系统上实施分子筛高效液相色谱(HPLC)。采用pH 6.8,0.1M磷酸钠,以1.0mL/min等度洗脱Phenomenex BioSep SEC-S3000柱(7.8×300mm)。
通过放射-TLC,采用ITLC-SG条(颇尔生命科学Pall Life Sciences,安阿伯,密歇根州)检测64Cu和111In标记产量,采用0.9%NaCl水溶液中的200mM EDTA洗脱,采用Bioscan 200成像扫描仪(Bioscan,华盛顿哥伦比亚特区)实施。在这些条件下,游离的放射性核素迁移Rf=0.9,而连接到四嗪6的放射性核素保持在原处。
使用Inveon临床前成像站(西门子医疗解决方案)获得PET/CT数据。
动物处理根据加利福尼亚大学,戴维斯分校,动物使用和管理委员会认可的方案处理所有动物。
统计学分析组变化描述为平均值±一标准偏差。采用双尾非配对t检验比较单组。P<0.05的组被认为是显著差异的。微软Excel版本12.8.9用于所有的统计计算。
实施例1 TCO改性海藻酸1的制备
以下描述TCO改性海藻酸的制备,首先制备TCO-连接体,然后连接至海藻酸。
(R,E)-N-(2-氨乙基)-2-(环辛-4-烯-1-基氧)乙酰胺(5)。化合物3(100mg,0.54mmol)、N-羟基琥珀酰胺(69mg,0.60mmol)和N,N'-二环己基碳二亚胺(123mg,0.60mmol)溶于CH2Cl2(2mL)并在室温搅拌2小时。通过PVDF膜滤掉沉淀脲。采用CH2Cl2(1mL)清洗膜。将母液加入到搅拌中的乙二胺(324mg,5.4mmol)的CH2Cl2(10mL)溶液。室温搅拌反应2小时。采用水(2×10mL)洗涤反应混合物。采用硫酸镁干燥有机层并减压浓缩。使用4:1的CH2Cl2和MeOH的混合物作为溶剂在制备型硅胶TLC上纯化产物。化合物5的产量是60mg(49%)。1H NMR(CDCl3)δ7.08(bs,1H),5.61-5.45(m,2H),3.92(q,J=11.23Hz,2H),3.64(dd,J1=9.91Hz,J2=4.73Hz,1H),3.36(q,J=6.07Hz,2H),2.87(bs,2H),2.31-2.16(m,4H),2.08-2.04(m,1H),1.89-1.49(m,5H)。13C NMR(CDCl3)δ170.34,135.48,131.37,75.80,68.31,41.28,40.05,34.28,32.53,29.74,27.85。HRMS:m/z[M+H]+计算C12H23N2O2227.1754,实测227.1791。
在标准碳化二亚胺化学条件下每克UP MVG海藻酸与176微摩TCO-胺结合,如之前对精氨酸-甘氨酸-天冬氨酸(RGD)和甘氨酸-组氨酸-赖氨酸(GHK)掺合所述。然后通过用含有降低盐浓度的去离子水透析4天纯化海藻酸产物,冷冻并冻干5-10天直到干燥。通过添加DPBS得到2.5%海藻酸溶液,并通过加入钙制备海藻酸凝胶。通过1H-NMR研究(见图20-22)证实海藻酸的共价修饰。不添加TCO的相同方案用于构建对照凝胶。用完全相同批次的TCO-凝胶完成体外和体内研究并在同一天使用以最大程度降低负载量或负载效率中的任何变化。
对于体外试验,800μl 2.5%的海藻酸溶液与200μl过饱和Ca(SO4)2溶液(每毫升双蒸水(ddH2O)0.21g Ca(SO4)2)。使用三通活塞混合溶液30秒从而获得2%终浓度海藻酸。混合物在特制塑料模具内的两块玻璃板之间胶凝并在室温孵育20分钟。总体具有与胶凝作用一致的均匀外观。用刮铲拾起盘片,单独称重。通常情况下,预制盘重约100毫克,并大致具有如下尺寸:8mm(直径)和2mm(高度)。
对于体内使用,2.5%的海藻酸凝胶溶液和过饱和硫酸钙溶液按上述相同的比例迅速混合,并立即以所需量注射到动物。
实施例2制备四嗪-改性诊断剂
如前所述合成Tz辐射探针2(R.Rossin,P.R.Verkerk,S.M.van den Bosch,R.C.Vulders,I.Verel,J.Lub,M.S.Robillard,Angew.Chem.Int.Ed.Engl.2010,49,3375).
实施例3制备(E)-环辛-2-烯基2-氨乙基氨基甲酸酯
根据Versteegen,R.M.等,Angew.Chem.Int.Ed.2013,52,14112-14116中描述的方案制备标题化合物。
实施例4制备可释放的TCO-改性诊断剂
将荧光素-NHS酯(134mg,0.283mmol)和(Z)-环辛-2-烯基2-氨乙基氨基甲酸酯(60.0mg,0.283mmol)溶于DMF(5mL)。加入三乙胺(77μL,0.566mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用1:9的MeOH:CH2Cl2混合物作为流动相进行纯化。产量=90mg(55.6%)。1H NMR(CD3OD,400MHz)δ8.42(s,1H),8.17(d,J=8.2Hz,1H),7.25(d,J=8.2Hz,1H),6.86(s,2H),6.57-6.50(m,4H),5.78(t,J=12.3Hz,1H),5.47(d,J=16.4Hz,1H),5.2(app s,1H),3.61-3.31(m,5H),2.32(bs,1H),1.98-1.88(m,3H),1.83-1.77(m,1H),1.69-1.04(m,5H),0.83-0.75(m,1H).13C NMR(CD3OD,100MHz)δ168.74,161.63,158.85,156.60,154.17,137.94,135.62,132.86,132.70,130.20,125.81,125.31,113.89,111.03,103.81,75.43,41.69,41.21,37.08,36.67,30.14,25.27.HRMS(ESI-MS)m/z:计算C32H30N2O8[M+H+]571.2080;实测571.2025.
实施例5制备TCO-改性诊断剂
将2-((E)-环辛-2-烯基氧)-N-(2-氨乙基)乙酰胺(50.0mg,0.221mmol)和荧光素-NHS酯(105mg,0.221mmol)溶于DMF(5mL)。加入三乙胺(60μL,0.442mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用1:9的MeOH:CH2Cl2混合物作为流动相进行纯化。产量=51mg(39.5%)。1H NMR(CD3OD,400MHz)δ8.46(s,1H),8.21(d,J=8.2Hz,1H),7.65(bs,1H),7.28(d,J=8.2Hz,1H),6.69(d,J=2.7Hz,2H),6.58-6.50(m,4H),5.46-5.44(m,2H),3.91(d,J=5.5Hz,2H),3.65-3.59(m,5H),2.67(s,1H),2.32-2.26(m,2H),2.32-2.26(m,2H),2.20-2.13(m,1H),2.08(d,J=4.2Hz,1H),1.96-1.92(m,1H),1.79-1.67(m,3H),1.54-1.46(m,1),1.30-1.25(m,1H),1.23-1.15(m,1H)。13C NMR(CD3OD,100MHz)δ173.60,170.67,168.69,154.21,137.21,137.66,136.66,136.75,132.53,130.30,125.89,125.09,113.84,110.98,103.79,77.56,69.38,41.31,40.75,40.07,39.94,35.50,33.58,30.82,29.05。HRMS(ESI-MS)m/z:计算C33H32N2O8[M+H+]585.2237;实测585.2183.
实施例6制备可释放的TCO-改性阿莫西林
将(E)-环辛-2-烯基4-硝基苯基碳酸酯(100mg,0.343mmol)和阿莫西林(82.0mg,0.224mmol)溶于DMF(5mL)。加入三乙胺(87μL,0.634mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用1:9的MeOH:CH2Cl2混合物作为流动相进行纯化。产量=36mg(31%)。1H NMR(CD3OD,400MHz)δ8.49(bs,1H),7.57(dd,J1=21.8Hz,J2=8.2Hz,1H),7.24(d,J=6.8Hz,2H),6.67(d,J=6.9Hz,2H),5.81–5.76(m,1H),5.51(d,J=16.4Hz,1H),5.29(s,1H),5.17(s,1H),4.92(s,1H),4.33(s,1H),3.62(s,3H),3.38(s,1H),2.50(s,2H),2.38(bs,1H),1.98-1.88(m,3H),1.81-1.75(m,1H),1.64-1.53(m,2H),1.37(s,3H),1.19-1.12(m,4H),0.81-0.78(m,2H)。13C NMR(CD3OD,100MHz)δ170.49,156.77,154.78,132.16,131.87,131.24,130.95,128.60,114.83,72.99,65.73,59.77,59.07,57.53,57.03,56.89,51.93,48.60,35.54,35.31,28.46,27.07,26.76,23.67,23.54。HRMS(ESI-MS)m/z:计算C33H32N2O8[M+MeO-]548.2072;实测548.2042。
实施例7制备TCO-改性阿莫西林
将2-((E)-环辛-2-烯基氧)乙酸(85.0mg,0.461mmol)、N-羟基琥珀酰胺(53.0mg,0.461mmol)和N,N′-二环己基碳二亚胺(95.0mg,0.461mmol)溶于CH2Cl2(5mL)。室温搅拌18小时。过滤沉淀物并减压浓缩上清液。加入阿莫西林(160mg,0.438mmol)的DMF(10mL)溶液并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用1:9的MeOH:CH2Cl2混合物作为流动相进行纯化。产量=20mg(8.6%)。1H NMR(CD3OD,400MHz)δ7.33(t,J=9.5Hz,1H),7.24(t,J=0.6Hz,1H),6.76(d,J=8.2Hz,2H),5.68-5.45(m,4H),4.32(bs,1H),3.99-3.09(m,2H),3.72(s,1H),3.47(bs,1H),2.67(s,6H),2.37-1.96(m,5H),1.85-1.80(m,3H),1.56(s,3H),1.47(s,3H),1.28-1.17(m,3H)。13C NMR(CD3OD,100MHz)δ175.08,172.65,171.82,158.81,131.16,131.07,130.69,130.13,129.98,129.89,116.69,83.19,68.67,66.71,59.88,58.86,57.05,53.13,35.26,35.14,34.26,34.19,27.18,26.75,26.69,26.41,23.44,23.41。HRMS(ESI-MS)m/z:计算C33H32N2O8[M+MeO-]548.2072;实测548.2042。
实施例8制备改性琼脂糖
NHS活化琼脂糖吸附柱购自皮尔斯/赛默飞世尔科技公司(Pierce/Thermo Fisher Scientific)(罗克福德,伊利诺斯州)。采用制造商推荐方案将图16B的1a或1b的胺前体连接至琼脂糖珠。简言之,33mg干琼脂糖与4μmol的1a或1b的胺前体在pH 7.4PBS缓冲液中孵育3小时,温和地混合。采用PBS洗涤琼脂糖珠数次。通过监测洗涤获得的上清液在520nm的吸光度估算结合到柱的1a或1b的量。琼脂糖上未反应的NHS基团被1M Tris(pH 7.4)封端。
实施例9捕获和释放连接体的动力学
用2微摩TCO-R-罗丹明((E)-5-((2-(((环辛-2-烯-1-基氧)羰基)氨基)乙基)氨甲酰基)-2-(3-(二甲基-4-偶氮烯)-6-(二甲基氨基)-3H-氧杂蒽-9-基)苯甲酸)处理采用上述方法的用1a或1b改性的琼脂糖珠2分钟。快速离心后收集上清,并将琼脂糖重悬于水中。在固定时间间隔收集上清并用ESI-MS分析。
在赛默飞世尔科技公司(西棕榈滩,加利福尼亚州)线性离子阱静电场轨道阱(LTQ Orbitrap Velos)质谱仪上采用石英毛细管发射器分析样本。为便于喷雾优化,在质谱分析之前将10%异丙醇加到各样本。以阳性模式分析释放产物,罗丹明乙二胺(5-((2-氨乙基)氨甲酰基)-2-(3-(二甲基-4-偶氮烯)-6-(二甲基氨基)-3H-氧杂蒽-9-基)苯甲酸)。单独合成释放产物罗丹明乙二胺用于ESI-MS校正(图SX)。采用校正曲线评估各步骤上清液中罗丹明乙二胺的量。
实施例10制备改性Tz-Me-海藻酸
在标准碳化二亚胺化学条件下每克UP MVG海藻酸与176微摩(4-(6-甲基-1,2,4,5-四嗪-3-基)苯基)甲胺(Tz-Me–胺)结合,如之前对RGD、GHK和TCO胺掺合所述。然后通过用含有降低盐浓度的去离子水透析4天纯化海藻酸产物,冷冻并冻干5-10天直到干燥。通过添加ddH2O得到2.5%海藻酸溶液,并通过加入钙制备海藻酸凝胶。通过1H-NMR研究(见图23)证实海藻酸的共价修饰。不添加TCO的相同方案用于构建对照凝胶。用完全相同批次的TCO-凝胶完成体外和体内研究并在同一天使用以最大程度降低负载量或负载效率中的任何变化。
对于体内使用,800微升2.5%的海藻酸溶液和200微升过饱和硫酸钙溶液(0.21g Ca(SO4)2/ml ddH2O)混合。采用三通活塞混合溶液30秒从而获得2%海藻酸终浓度。立即以所需量将混合物注射给动物。
实施例11捕获和释放连接体的体内评价
IACUC批准后,通过不注射或注射一种海藻酸(对照相对Tz-凝胶)在nu/nu小鼠上实施体内荧光实时生物分布研究。然后对象接受尾静脉注射TCO-R-F或TCO-NR-F。阴性对照为:1.无凝胶,用TCO-R-F(阴性对照,小鼠1);2.含TCO-R-F的对照海藻酸(凝胶对照,小鼠4)。两实验组为Tz-凝胶以及或者TCO-R-F(释放方案,小鼠2)或TCO-NR-F(固定化方案,小鼠3)。采用IVIS光谱仪(珀金埃尔默,马萨诸塞州)检测荧光并记录辐射。
实施例12可释放万古霉素的最小抑制浓度(MIC)
我们在ddH2O中过夜制备含常规海藻酸凝胶或Tz-凝胶的万古霉素或TCO-R-万古霉素的连续稀释液。翌日,将2.2%Mueller Winto肉汤中的发光甲氧西林敏感金黄色葡萄球菌(Staph.Aureus,MSSA,Xen 29,珀金埃尔默,MA)加至混合物。然后将培养板置入培养箱24小时,并使其生长24小时(n=3)。然后采用IVIS光谱仪(珀金埃尔默,马萨诸塞州)检测荧光并记录辐射率。
实施例13制备可释放的TCO-罗丹明
如Brunet,A.;Aslam,T.;Bradley,M.Bioorg.Med.Chem.Lett.2014,24,3186-3188所描述的合成罗丹明-NHS酯。将罗丹明-NHS酯(50mg,0.095mmol)和(E)-环辛-2-烯基-2-氨乙基氨基甲酸酯(40.0mg,0.190mmol)溶于CH2Cl2(5mL)。加入三乙胺(129μL,0.95mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。采用7.5:2.5:90MeOH:Et3N:CH2Cl2混合物作为流动相通过制备型薄层色谱纯化。产量=28mg(47%)。1H NMR(CD3OD,400MHz)δ8.54(s,1H),8.04(d,J=8.2Hz,1H),7.34(d,J=8.2Hz,1H),7.23(d,J=9.6Hz,1H),6.99(dd,J1=2.7Hz,J2=9.5Hz,1H),6.89(d,J=2.8Hz,1H),5.85(t,J=13.7Hz,1H),5.55(d,J=16.4Hz,1H),5.26(s,1H),3.67-3.36(m,4H),3.32-3.21(m,9H),2.93-2.77(m,6H),2.49-2.36(m,1H),2.10-1.79(m,5H),1.78-1.41(m,4H),1.39-1.25(m,1H),1.22-1.06(m,10H),0.93-0.79(m,1H).13C NMR(CD3OD,100MHz)δ172.47,169.41,161.68,159.08,158.76,142.06,137.19,132.96,132.70,130.90,129.85,129.63,115.09,114.86,97.53,76.85,75.38,41.96,41.78,41.01,37.18,36.92,30.21,25.35。HRMS(ESI)m/z:计算C36H41N4O6[M+1]+625.3026;实测625.2976.
实施例14制备非-可释放的TCO-罗丹明
将罗丹明-NHS酯(50mg,0.095mmol)和2-((E)-环辛-4-烯基氧)-N-(2-氨乙基)乙酰胺(43.0mg,0.190mmol)溶于CH2Cl2(5mL)。加入三乙胺(129uL,0.95mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用7.5:5:2.5:90的MeOH:Et3N:CH2Cl2混合物作为流动相进行纯化。产量=35mg(58%).1H NMR(CD3OD,400MHz)δ8.57(s,1H),8.07(d,J=8.2Hz,1H),7.34(d,J=8.2Hz,1H),7.22(d,J=8.2Hz,2H),6.97(dd,J1=2.7Hz,J2=9.6Hz,2H),6.88(d,J=2.7Hz,2H),5.61-‐5.43(m,2H),3.94(d,J=4.1Hz,2H),3.69-‐3.55(m,6H),3.31(s,2H),2.41-‐2.31(m,2H),2.27-‐2.14(m,2H),2.03-‐1.95(m,1H),1.90(s,2H),1.84-1.71(m,4H),1.61-‐1.50(m,2H).13C NMR(CD3OD,100MHz)δ173.49,172.35,169.46,161.47,159.04,158.71,142.12,136.97,136.88,132.70,132.57,129.84,129.65,115.02,114.83,97.55,77.65,69.44,41.40,41.00,35.58,33.62,30.88,29.10.HRMS(ESI)m/z:计算C37H43N4O6[M+1]+639.3183;实测639.3151.
实施例15罗丹明释放产物
将罗丹明-NHS酯(20.0mg,0.0379mmol)和N-Boc-乙二胺(13.2mg,0.0758mmol)溶于CH2Cl2(5mL)。加入三乙胺(51L,0.379mmol)并在室温搅拌18h。高真空下蒸发溶剂并将反应混合物重溶于甲醇。通过制备型薄层色谱采用10:5:85的MeOH:Et3N:CH2Cl2混合物作为流动相进行纯化。分离产物用4:1的CH2Cl2:TFA(5mL)混合物处理1h。产量=18mg(81%)。1H NMR(CD3OD,400MHz).8.83(s,1H),8.32(d,J=9.6Hz,1H),7.55(d,J=8.1Hz,1H),7.13(d,J=9.6Hz,2H),7.05(dd,J1=6.8Hz,J2=2.7Hz,2H),6.96(d,J=2.7Hz,2H),3.77(t,J=5.4Hz,,2H),3.30(s,12H),3.22-3.17(m,2H)。13C NMR(CD3OD,100MHz).169.28,167.47,160.69,159.10,138.52,132.63,132.11,132.02,131.67,115.71,114.85,97.63,41.08,40.97,39.06。HRMS(ESI)m/z:计算C27H29N4O4[M+1]+473.2189;实测473.2141.
实施例16可释放的TCO-万古霉素
将万古霉素(50.0mg,0.0345mmol)和三乙胺(20μL,0.145mmol)溶于水(1mL)中。加入(E)-环辛-2-烯基4-硝基苯基碳酸酯(19.0mg,0.0652mmol)的DMF(25μL)溶液。在40℃搅拌反应混合物18h。通过45μm PTFE膜滤掉沉淀。上清液经HPLC纯化从而获得标题化合物。梯度10-60%CH3CN的水溶液用于HPLC纯化。产量=3mg(5.5%)。1H NMR(CD3OD,400MHz)δ7.61(s,1H),7.55-7.38(m,2H),7.19(bs,1H),7.03(s,1H),6.81(bs,1H),6.44(s,1H),6.38(s,1H),6.05(bs,1H),5.55-5.37(m,2H),5.32(s,1H),5.23(s,1H),4.68(s,36H),4.52-4.31(m,2H),4.15(s,1H),4.00(t,J=8.2Hz,1H),3.85-3.61(m,3H),3.56(t,J=8.2Hz,1H),3.35(s,1H),2.67(s,4H),2.03-1.88(m,2H),1.75-1.39(m,3H),1.33(s,3H),1.05(s,3H),0.75(dd,J1=8.2Hz,J2=13.7Hz,6H)。HRMS(ESI)m/z:计算C75H87Cl2N9O26[M+1]+1602.4660;实测1602.5256.
实施例17可释放的TCO-达托霉素
达托霉素(100mg,0.062mmol)溶于水(1mL)中。加入(E)-环辛-2-烯基4-硝基苯基碳酸酯(36mg,0.123mmol)的DMF(50μL)溶液,随后加入三乙胺(167mg,1.65mmol)。室温搅拌反应混合物18小时。使用半制备型菲罗门(Phenomenex)Luna 5u C18(2)柱和梯度10-65%CH3CN的水溶液经HPLC纯化后获得标题产物,为白色泡沫。产量=44mg(40%)。1H NMR(CD3OD,400MHz)δ7.66(bs,2H),7.60-7.46(m,2H),7.39(s,1H),7.25-7.11(m,2H),7.02(s,1H),6.96(s,1H),6.83-6.78(m,1H),6.56(s,1H),6.00-5.68(m,4H),5.68-5.57(m,1H),5.54(s,1H),5.51-5.26(m,8H),5.14(bs,1H),4.96(bs,1H),4.67(s,1H),4.64(s,1H),4.58(s,1H),4.14(s,1H),3.88-3.79(m,2H),3.78-3.71(m,1H),3.59-3.43(m,2H),3.19(q,J=8.2Hz,7H),2.95(bs,4H),2.59-2.32(m,5H),2.23-1.76(m,15H),1.56-1.44(m,8H),1.30(t,J=8.1Hz,10H),1.23-1.16(m,5H),1.00-0.87(m,9H)。MS(ESI)m/z:计算C75H87Cl2N9O26[M+H+MeCN]+1813.82;实测1813.80.
实施例18可释放的TCO-阿霉素
根据Versteegen,R.M.等.,Angew.Chem.Int.Ed.2013,52,14112-14116所描述合成(E)-环辛烯阿霉素偶联物。1H NMR(CDCl3)谱图与公开数据相符。
实施例19可释放的TCO-环腺苷酸
环腺苷酸(80mg,0.243mmol)和(E)-环辛-2-烯基4-硝基苯基碳酸酯(142mg,0.486mmol)溶于无水DMF(8mL)。加入4-二甲基氨基吡啶(238mg,1.94mmol)并在30℃搅拌反应混合物18h。高真空下去除溶剂,采用15%MeOH的二氯甲烷溶液作为流动相经制备柱色谱后获得标题产物,为白色泡沫。产量=45mg(38%)。1H NMR(CDCl3,400MHz)δ7.61(s,1H),7.57-7.41(m,2H),7.19(bs,1H),7.03(s,1H),6.81(bs,1H),6.44(s,1H),6.38(s,1H),6.06(bs,1H),5.53-5.39(m,2H),5.32(s,2H),5.24(s,1H),4.48(bs,2H),4.16(s,1H),4.00(t,J=6.8Hz,1H),3.83-3.61(m,3H),3.60-3.50(m,1H),3.35(s,1H),2.68(s,4H),2.03-1.90(m,2H),1.75-1.40(m,3H),1.33(s,3H),1.05(s,3H),0.74(dd,J1=8.2Hz,J2=13.6Hz,6H)。HRMS(ESI)m/z:计算C19H25N5O8P[M+1]+482.1441;实测482.1461.
实施例20可释放的TCO-化合物3
化合物3(132mg,0.413mmol)和(E)-环辛-2-烯基4-硝基苯基碳酸酯(80mg,0.275mmol)溶于无水DMF(5mL)。加入三乙胺(167mg,1.65mmol)并在室温搅拌反应混合物18小时。高真空下去除溶剂,采用10%MeOH的二氯甲烷溶液作为流动相经制备柱色谱后获得标题产物,为白色泡沫。产量=70mg(36%)。1H NMR(CD3OD,400MHz)δ8.28(s,1H),5.93(d,J=6.9Hz,1H),5.89-5.78(m,1H),5.54(bs,1H),5.28(bs,1H),4.73(t,J=5.5Hz,1H),4.37-4.31(m,1H),4.23-4.13(m,3H),3.92(d,J=10.9Hz,1H),3.73(d,J=12.3Hz,1H),3.65-3.40(m,1H),3.35(s,1H),3.31(s,1H),2.47-2.30(m,1H),2.10-1.75(m,4H),1.75-1.57(m,2H),1.53(m,1H),1.26(bs,1H),1.18-1.06(m,2H),0.91-0.74(m,1H)。HRMS(ESI)m/z:计算C22H28N6O6[M+1]+473.2149;实测473.2117.
尽管为了清楚理解的目的以说明和举例的方式相当详细描述前述发明,但本领域的技术人员将理解,在所附权利要求的范围内可以作出某些改变和修改。另外,本文提供的各参考通过引用以其全部内容并入本文,如同通过引用单独并入各参考相同的程度。本申请和本文提供的参考之间存在冲突之处,以本申请为准。
- 还没有人留言评论。精彩留言会获得点赞!