一种暖胃舒乐片及其制备方法与检测方法与用途与流程
本发明涉及中药领域,具体涉及一种暖胃舒乐片药物及其制备方法。
背景技术:
暖胃舒乐片,由黄芪、大红袍、延胡索、白芍等药物制成,其具有温中补虚,调和肝脾,行气活血,止痛生肌的功效。用于脾胃虚寒及肝脾不和型慢性胃炎症见脘腹疼痛,腹胀喜温,反酸嗳气。
该品种年销售额达2多亿元,按照中央“供给侧结构性改革”的精神要求,专利申请人在现有品种的基础上,对该品种进行了“传统名优中药二次开发”的研究工作。开发重在工艺改造、质量标准的提高和有效物质基础确定以提高有效性等方面。
暖胃舒乐片现有的制备方法,是采取水提取的制备方法,专利申请人在对此药物的制备工艺进行持续多年的研究中发现,现有的制备工艺存在诸多问题,例如对水溶性成分的提取,对挥发性成分的提取,都是不足的,申请人在进行大量的、反复的实验研究后,对现有的制备工艺进行了改进提高,得到了本发明的制备工艺,该制备工艺可以得到了该药物更多的活性成分,更有利于提高药效,尤其是对于晕动症的治疗,疗效的提高更加显著。同时,本发明还提供了新的药物检测方法,能够更加有效地控制药物的品质。
技术实现要素:
本发明的目的是提供一种暖胃舒乐片的药物。
本发明的另一目的是提供该药物的制备方法。
本发明还提供了该药物的检测方法。
本发明还提供了该药物的制药用途。
本发明的目的是由以下方式实现的:
一种暖胃舒乐片药物,该药物是由以下重量份原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份。
所述的暖胃舒乐片药物,该药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得。
一种暖胃舒乐片药物的检测方法,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份;该暖胃舒乐片药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得;
可以采用如下高效液相法方法对其进行指纹图谱的检测:
(1)色谱条件:色谱柱Crosmosil C18柱,流动相以乙腈为流动相A,以1%冰醋酸溶液为流动相B,梯度洗脱:0min至15min,流动相A从10%线性增加至25%,流动相B从90%线性下降至75%,16min至30min,流动相A从25%线性增加至70%,流动相B从75%线性下降至30%,31min至50min,流动相A保持在70%,流动相B保持在30%,检测波长228nm,流速1.5mL·min-1;柱温25℃,进样体积10μL;
(2)对照品溶液的制备:精密称取熊竹素对照品、4-苯醌对照品、芍药新苷甲素对照品,置量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液;
(3)供试品溶液的制备:精密量取药物样品,精密加入甲醇,称重,超声提取,离心,取出,放冷,补重,摇匀,微孔滤膜滤过,取续滤液,即得供试品溶液;
(4)测定:精密吸取对照品溶液和供试品溶液注入高效液相色谱仪,进行测定。
所述的暖胃舒乐片药物的检测方法,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份,该暖胃舒乐片药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得;
优选采用如下高效液相法方法对其进行指纹图谱的检测:
(1)色谱条件:色谱柱Crosmosil C18柱,流动相以乙腈为流动相A,以1%冰醋酸溶液为流动相B,梯度洗脱:0min至15min,流动相A从10%线性增加至25%,流动相B从90%线性下降至75%,16min至30min,流动相A从25%线性增加至70%,流动相B从75%线性下降至30%,31min至50min,流动相A保持在70%,流动相B保持在30%,检测波长228nm,流速1.5mL·min-1;柱温25℃,进样体积10μL;
(2)对照品溶液的制备:精密称取熊竹素对照品4.13mg、4-苯醌对照品4.21mg、芍药新苷甲素对照品2.15mg,置10mL量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液;
(3)供试品溶液的制备:精密量取药物样品0.5g,精密加入甲醇25mL,称重,超声提取30min,取出,放冷,补重,摇匀,0.45μm微孔滤膜滤过,取续滤液,即得供试品溶液;
(4)测定:精密吸取对照品溶液和供试品溶液各10μL注入高效液相色谱仪,进行测定。
一种暖胃舒乐片药物的检测方法,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份;该暖胃舒乐片药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得;
采用高效液相色谱法测定药物中消旋-丁香树脂酚的含量:
(1)色谱条件:采用HypersilDs,色谱柱;流动相:比例为70~90:20的乙腈-0.05%磷酸溶液;检测波长:260~280nm;柱温:30~40℃;流速:0.5~2.0mL·min-1;
(2)对照品溶液的制备:精密称取干燥至恒重的消旋-丁香树脂酚对照品,加甲醇制成对照品溶液;
(3)供试品溶液制备:精密称取药物样品,加甲醇,加热回流,提取液回流溶剂并浓缩至干,残渣加水溶解,用水饱和的正丁醇振摇提取,合并正丁醇提取液,用氨试液洗涤,正丁醇提取液回收溶剂至干,残渣加甲醇溶解并转移至容量瓶中,加甲醇至刻度,摇匀,滤过,取滤液,即得供试品溶液;
(4)测定:精密称取对照品溶液和供试品溶液各5~20μL,注入高效液相色谱仪进行测定。
上述的暖胃舒乐片药物的检测方法,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份,该暖胃舒乐片药物优选采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得;
优选采用高效液相色谱法测定药物中消旋-丁香树脂酚的含量:
(1)色谱条件:采用HypersilDs色谱柱;规格:4.0mm×125mm,5μm,流动相:比例为80:20的乙腈-0.05%磷酸溶液;检测波长:268nm;柱温:35℃;流速:1.0mL·min-1;
(2)对照品溶液的制备:精密称取80℃干燥至恒重的消旋-丁香树脂酚对照品适量,加甲醇制成每1mL含0.2mg的溶液,即得对照品溶液;
(3)供试品溶液制备:精密称取药物样品10g,加甲醇40mL,加热回流4h,提取液回流溶剂并浓缩至干,残渣加水10mL溶解,用水饱和的正丁醇振摇提取5次,每次20mL,合并正丁醇提取液,用氨试液洗涤3次,每次15mL,正丁醇提取液回收溶剂至干,残渣加甲醇溶解并转移至10mL容量瓶中,加甲醇至刻度,摇匀,滤过,取滤液,即得供试品溶液;
(4)测定:精密称取对照品溶液和供试品溶液各10μL,注入高效液相色谱仪进行测定。
一种暖胃舒乐片药物在制备治疗晕动症药物中的应用,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份,该暖胃舒乐片药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得;
采用如下高效液相法方法对药物进行指纹图谱检测:
(1)色谱条件:色谱柱Crosmosil C18柱,流动相以乙腈为流动相A,以1%冰醋酸溶液为流动相B,梯度洗脱:0min至15min,流动相A从10%线性增加至25%,流动相B从90%线性下降至75%,16min至30min,流动相A从25%线性增加至70%,流动相B从75%线性下降至30%,31min至50min,流动相A保持在70%,流动相B保持在30%,检测波长228nm,流速1.5mL·min-1;柱温25℃,进样体积10μL;
(2)对照品溶液的制备:精密称取熊竹素对照品4.13mg、4-苯醌对照品4.21mg、芍药新苷甲素对照品2.15mg,置10mL量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液;
(3)供试品溶液的制备:精密量取药物样品0.5g,精密加入甲醇25mL,称重,超声提取30min,取出,放冷,补重,摇匀,0.45μm微孔滤膜滤过,取续滤液,即得供试品溶液;
(4)测定:精密吸取对照品溶液和供试品溶液各10μL注入高效液相色谱仪,进行测定;
采用如下高效液相色谱法测定药物中消旋-丁香树脂酚的含量:
(1)色谱条件:采用HypersilDs色谱柱;规格:4.0mm×125mm,5μm,流动相:比例为80:20的乙腈-0.05%磷酸溶液;检测波长:268nm;柱温:35℃;流速:1.0mL·min-1;
(2)对照品溶液的制备:精密称取80℃干燥至恒重的消旋-丁香树脂酚对照品适量,加甲醇制成每1mL含0.2mg的溶液,即得对照品溶液;
(3)供试品溶液制备:精密称取药物样品10g,加甲醇40mL,加热回流4h,提取液回流溶剂并浓缩至干,残渣加水10mL溶解,用水饱和的正丁醇振摇提取5次,每次20mL,合并正丁醇提取液,用氨试液洗涤3次,每次15mL,正丁醇提取液回收溶剂至干,残渣加甲醇溶解并转移至10mL容量瓶中,加甲醇至刻度,摇匀,滤过,取滤液,即得供试品溶液;
(4)测定:精密称取对照品溶液和供试品溶液各10μL,注入高效液相色谱仪进行测定。
一种暖胃舒乐片药物在制备治疗扁桃体炎药物中的应用,该暖胃舒乐片药物是由以下重量份的原料制成的:黄芪113重量份、大红袍45重量份、延胡索45重量份、白芍113重量份、鸡矢藤13重量份、白及113重量份、砂仁6重量份、五倍子6重量份、肉桂11重量份、丹参45重量份、甘草45重量份、炮姜45重量份,该暖胃舒乐片药物采用如下方法制备:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂,即得。
实验一:本发明药物治疗晕动病的实验研究
发明人对犬进行了治疗晕动病的实验研究。
1 材料与方法
1.1 实验材料
1.1.1 实验动物
家养健康犬6只,雌雄各半,体重5~6kg,普通级,由广州中医药大学实验动物中心提供,动物生产许可证号:SCXK(粤)2008-0020。
1.1.2 药品
1.1.2.1 本发明药物:处方:黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g;
制备方法:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂1000片,即得。
1.1.2.2 对比药物:处方:黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g;
制备方法:以上十二味,肉桂、砂仁、鸡矢藤、炮姜粉碎成细粉,其余黄芪等八味加水煎煮三次,第一、二次各1.5h,第三次1h,滤过,合并滤液,浓缩成稠膏状,加入上述细粉,混匀,干燥,粉碎成细粉,制成颗粒,干燥,压制成100片,即得。
1.1.3 设备
旋转刺激装置,由广州中医药大学药学院按Crampton等报道(Crampton GH,Lucot JB. A stimulator for laboratory studies of motion sickness in cars [J].Aviat Space Environ Med,1985,56:462.)仿制而成。该装置旋转轮直径1.2m,配有透明通气的动物箱及转速控制钮。根据转速及旋转轮的直径,可计算出角加速度的大小(即诱发运动病发作的刺激量)。
1.2 方法
1.2.1 动物筛选
实验时,将实验犬无束缚地置入旋转装置的动物箱中,绕水平轴顺时针旋转,以70°·(s)2-1角加速度加速4.5s,达最大速度315°·(s)-1后匀速旋转3s,立即以126°·(s)2-1角减速度减速,直至旋转停止为1个周期,如此循环进行可诱发晕动病(姜正林.生姜合剂对运动病大鼠血液肾素、血管紧张素、醛固酮水平的影响 [J].中华航海医学与高气压医学杂志 , 2001, 8( 1):16)。
旋转开始后半小时内,被选犬若出现流诞或呕吐症状者,即被视为对本试验敏感,作为本实验用犬,共6只,然后分别标记编号,按常规适应性饲养2周。
1.2.2 自身对照试验
第1次进行本发明药物治疗组(观察组)实验。每犬于开始实验前半小时内,灌胃本发明药物3片,然后将犬置于动物箱中旋转,测定实验开始至动物出现流诞或呕吐症状的时长,并分别记录。
实验结束后,然后按常规饲养10天,再进行第2次实验。
第2次进行对比药物对照组实验。每犬于开始实验前半小时内,灌胃本发明药物3片,然后将犬置于动物箱中旋转,测定实验开始至动物出现流诞或呕吐症状的时长,并分别记录。
2 结果
实验观察组与对照组在接受相同旋转刺激量后出现呕吐或流涎的时长见表1。观察组诱导犬产生流涎或呕吐症状的时间较对照组明显延长,经配对t检验,两组差异有极显著性(P<0.01,t=4.9035),提示观察组的疗效明显优于对照组。
表1 两组实验犬出现呕吐或流涎的时间(min)
3 讨论与结论
本实验通过在相同的旋转刺激量条件下,比较观察组与对照组诱导恶心呕吐的时间,时间越长,动物对旋转刺激的耐受性越强,表明疗效越佳。结果表明,本发明药物较对比药物有更强的抑制胃肠道植物神经功能紊乱的作用,对防治晕动病的发作疗效确切。这也充分说明,本发明提供的独特的制备方法,显著地提高了药物的作用疗效。
实验二:本发明药物的HPLC指纹图谱研究
采用高效液相法建立本发明药物的指纹图谱检测分析方法。
1 材料
1.1 仪器
Accela高效液相色谱仪(上海赛默飞世尔科技上海有限公司);PDA检测器,四元梯度泵,软件为Empower2数据处理软件系统;电子天平(上海赛多利斯贸易有限公司);KQ-300DE数控超声波清洗器(上海凌科实业发展有限公司)。
1.2 试药
本发明药物,10批,参照本发明说明书具体实施方式中的实施例1制备,见表13。对照品熊竹素、4-苯醌、芍药新苷甲素购于上海极威生物科技有限公司生产。乙腈为色谱纯,液相用水为屈臣氏蒸馏水,其余试剂均为分析纯。
表2 本发明药物测试样品批号
2 方法与结果
2.1 色谱条件
色谱柱C18柱,规格:4.6mm×250mm,5μm,流动相以乙腈为流动相A,以1%冰醋酸溶液为流动相B,按表14进行梯度洗脱,检测波长228nm,流速1.5mL·min-1;柱温25℃,进样体积10μL。
表3 流动相洗脱条件
2.2 对照品溶液的制备
精密称取熊竹素对照品4.13mg、4-苯醌对照品4.21mg、芍药新苷甲素对照品2.15mg,置10mL量瓶中,加甲醇溶解并稀释至刻度,即得。
2.3 供试品溶液的制备
上述S1~S10号药物样品,精密量取药物样品0.5g,精密加入甲醇25mL,称重,超声提取30min,取出,放冷,补重,摇匀,0.45μm微孔滤膜滤过,取续滤液作为供试品溶液。
2.4 方法学考察
2.4.1 重复性试验
取同一批号样品,分别精密称取6份,按照2.3项下方法制备供试品溶液,分别进样,以4号峰熊竹素为参照峰,计算出1~18号共有指纹峰的相对保留时间和相对峰面积的RSD<3%,同时用相似度评价软件计算各色谱指纹图谱的相似度均大于0.99,表明方法重复性良好。
2.4.2 精密度试验
取重复性试验中同一供试品溶液,连续进样5次,以4号峰熊竹素为参照峰,计算出1~18号共有指纹峰的相对保留时间和相对峰面积的RSD均小于3%,同时用相似度评价软件计算各色谱指纹图谱的相似度均大于0.99,表明仪器稳定,精密度良好。
2.4.3 稳定性试验
取重复性试验中同一供试品溶液,分别在0,4,8,12,24h进样,以4号峰熊竹素为参照峰,计算出1~18号共有指纹峰的相对保留时间和相对峰面积的RSD均小于3%,同时用相似度评价软件计算各色谱指纹图谱的相似度均大于0.99,表明供试品溶液在24h内稳定。
2.5 指纹图谱的建立
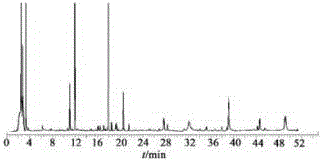
精密吸取2.2项下的对照品溶液及2.3项下制备的供试品溶液各10μL,注入高效液相色谱仪,得到10个批号本发明药物HPLC指纹图谱,结果见图1。
通过与对照品的比较,确认3号峰为芍药新苷甲素,4号峰为熊竹素,7号峰为4-苯醌,其中4号峰熊竹素峰面积较大且较稳定,故选择此峰为参照峰。将10批本发明药物指纹图谱导入采用药典委员会推荐的中药色谱指纹图谱相似度评价系统(A版),进行色谱峰匹配,匹配结果见图2,所得对照指纹图谱见图3。
以4号峰熊竹素为参照,去除溶剂峰后确定18个共有峰为构成本发明药物指纹图谱的特征峰,样品共有峰的相对保留时间及相对峰面积分别列于表4和表5。10批本发明药物与对照指纹图谱相似度计算结果分别为0.968,0.999,0.998,0.998,0.997,0.990,0.990,0.999,0.999,0.998。
表4 10批本发明药物的共有峰的相对保留时间
表5 10批本发明药物共有峰的相对峰面积
3 讨论
以熊竹素、4-苯醌、芍药新苷甲素为对照峰确定了本发明药物的18个共有峰,并通过相似度计算软件得到10批本发明药物的对照图谱及各批样品与对照图谱之间的相似度,结果均大于0.9,表明本发明药物的制备工艺稳定。对照图谱能提供较为全面的药品信息,可作为本发明药物的品质控制与评价方法之一。
实验三:高效液相色谱法测定本发明药物中消旋-丁香树脂酚的含量
1仪器与试药
1.1仪器
Accela高效液相色谱仪(上海赛默飞世尔科技上海有限公司)。
1.2试药
消旋-丁香树脂酚对照品(上海极威生物科技有限公司生产);甲醇(色谱醇,上海生化工助剂厂生产);其他试剂为分析纯。
2方法与结果
2.1处方
黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g;
2.2 制备方法
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂1000片,即得。
2.3 消旋-丁香树脂酚的含量测定
2.3.1 HPLC色谱条件
采用HypersilDs,规格:4.0mm×125mm,5μm,色谱柱;流动相:比例为80:20的乙腈-0.05%磷酸溶液;检测波长:268nm;柱温:35℃;流速:1.0mL·min-1;进样量:10μL;在该色谱条件下,对照品及样品色谱峰良好,无肉桂阴性对照对测定无干扰。
2.3.2对照品溶液的制备
精密称取80℃干燥至恒重的消旋-丁香树脂酚对照品适量,加甲醇制成每1mL含0.2mg的溶液,即得对照品溶液;
2.3.3供试品溶液与阴性对照液的制备
精密称取本发明药物10g,加甲醇40mL,加热回流4h,提取液回流溶剂并浓缩至干,残渣加水10mL溶解,用水饱和的正丁醇振摇提取5次,每次20mL,合并正丁醇提取液,用氨试液洗涤3次,每次15mL,正丁醇提取液回收溶剂至干,残渣加甲醇溶解并转移至10mL容量瓶中,加甲醇至刻度,摇匀,滤过,取滤液即得;另按处方比例制备不含肉桂的阴性对照品,同法制成阴性对照液。
2.3.4标准曲线的绘制
精密称取80℃干燥至恒重的消旋-丁香树脂酚对照品适量,用甲醇制成10.4,20.8,41.6,83.2,166.4μgmL-1系列浓度的溶液,分别精密量取上述5种浓度溶液各10μL,注入高效液相色谱仪进行测定。
以峰面积比值与浓度进行线性回归,得回归方程为:A=21.2763C — 0.1391,r=0.9999。表明消旋-丁香树脂酚在10.4~166.4μgmL-1浓度范围内呈良好的线性关系。
2.3.5稳定性试验
精密吸取供试品溶液10μL,分别于0、1、2、4、8h进样,并计算消旋-丁香树脂酚含量。结果8h内RSD为0.45%(n=5)。表明样品溶液在8h内稳定。
2.3.6重复性试验
按供试品溶液制备方法平行处理样品5份,依法测定消旋-丁香树脂酚含量并计算。结果测得消旋-丁香树脂酚平均含量为0.12mg·g-1,RSD为1.3%。
2.3.7精密度实验
精密吸取消旋-丁香树脂酚对照品溶液,重复进样5次,依法测定峰面积。结果RSD为0.23%(n=5)。表明精密度较好。
2.3.8回收率实验
精密称取已知消旋-丁香树脂酚含量的同一批号的样品6份,按高、中、低浓度分别精密加入适量的消旋-丁香树脂酚对照品溶液,按样品测定项下操作,依法测定,计算回收率。结果平均回收率为100.3%,RSD为0.45%(n=5)。
2.3.9样品含量测定
分别量取对照品溶液和供试品溶液适量,用微孔滤膜滤过,各进样10μL,按上述色谱条件测定3批样品,平行测定5次。按外标法以峰面积计算供试品溶液消旋-丁香树脂酚的含量。本品含消旋-丁香树脂酚应为标示含量的95%~105%,以每1g样品含消旋-丁香树脂酚计,不得少于0.12mg。3批样品含量分别为100.8%(RSD=1.2%),101.7%(RSD=1.3%),99.2%(RSD=1.1%)。
实验四:不同药物中消旋-丁香树脂酚的含量测定
1 待测样品
1.1 本发明药物:
1.1.1 处方
黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g;
1.1.2 制备方法
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂1000片,即得。
1.2 对比药物
1.2.1 处方
黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g。
1.2.2 制备方法
以上十二味,肉桂、砂仁、鸡矢藤、炮姜粉碎成细粉,其余黄芪等八味加水煎煮三次,第一、二次各1.5h,第三次1h,滤过,合并滤液,浓缩成稠膏状,加入上述细粉,混匀,干燥,粉碎成细粉,制成颗粒,干燥,压制成100片,即得。
2 测定方法
采用本发明实验三提供的高效液相色谱法测定各个药物中的消旋-丁香树脂酚的含量。
3 测定结果
测定结果见表6
表6 样品含量测定结果
4 讨论与结论
从上表中可以看出,本发明药物中的消旋-丁香树脂酚的含量明显高于对比药物,这可能也是本发明药物在治疗晕动症方面的效果优于其他药物的原因。
附图说明:
图1:本发明暖胃舒乐片药物 HPLC指纹图谱,其中图A为对照品HPLC色谱图; 图B为供试品HPLC色谱指纹图谱;
图2:10 批样品匹配色谱
图3:对照品指纹图谱
具体实施方式:
实施例1:本发明药物
处方:处方:黄芪113g、大红袍45g、延胡索45g、白芍113g、鸡矢藤13g、白及113g、砂仁6g、五倍子6g、肉桂11g、丹参45g、甘草45g、炮姜45g;
制备方法:
(1)将1/2处方量的砂仁、1/2处方量的鸡矢藤混合,粉碎成细粉,得混合细粉A,备用;
(2)取肉桂、炮姜、延胡索,将其进行水蒸气蒸馏提取,从中提取挥发油B,备用;
(3)将肉桂、炮姜、延胡索水蒸气蒸馏提取后的水溶液过滤,得水蒸气蒸馏后的水溶液,加热浓缩,得水蒸气蒸馏后的水浓缩液C,备用;保留水蒸气蒸馏提取后的药渣,备用;
(4)将步骤(3)所得水蒸气蒸馏提取后的药渣与黄芪、大红袍、白芍、白及、五倍子、丹参、甘草、1/2处方量的砂仁、1/2处方量的鸡矢藤混合,向其中加3倍量的90%乙醇回流提取二次,每次回流提取2小时,合并乙醇提取液,滤过,得混合乙醇提取液,回收乙醇并浓缩,得混合乙醇提取浓缩液,备用;保留乙醇提取后的药渣,备用;
(5)将步骤(4)所得混合乙醇提取浓缩液经D101B型大孔吸附树脂处理,用柱体积8倍量的pH 4的水溶液进行洗脱水溶液进行洗脱,弃去洗脱液,再用柱体积8倍量的50%的丙酮水溶液进行洗脱,收集洗脱液,然后在70℃下蒸馏,除去丙酮,得到提取液D;
(6)将步骤(4)所得乙醇提取后的药渣加水煎煮三次,第一、二次各1.5小时,第三次1小时,滤过,合并滤液,得混合水提取液,加热浓缩,得混合水提取浓缩液;
(7)将步骤(6)所得混合水提取浓缩液经聚酰胺树脂柱处理,用柱体积2~5倍的乙酸乙酯洗脱,收集洗脱液,然后在70℃下蒸馏,除去乙酸乙酯,得到提取液E;
(8)将步骤(5)所得提取液D、步骤(7)所得提取液E、步骤(3)所得水蒸气蒸馏后的水浓缩液C合并,加入步骤(1)所得混合细粉A,再加入步骤(2)所得挥发油B,加入淀粉,混匀,制粒,压片,包衣,制成片剂1000片,即得。
检测方法:
1、采用如下高效液相法方法对其进行指纹图谱的检测:
(1)色谱条件:色谱柱Crosmosil C18柱,流动相以乙腈为流动相A,以1%冰醋酸溶液为流动相B,梯度洗脱:0min至15min,流动相A从10%线性增加至25%,流动相B从90%线性下降至75%,16min至30min,流动相A从25%线性增加至70%,流动相B从75%线性下降至30%,31min至50min,流动相A保持在70%,流动相B保持在30%,检测波长228nm,流速1.5mL·min-1;柱温25℃,进样体积10μL;
(2)对照品溶液的制备:精密称取熊竹素对照品4.13mg、4-苯醌对照品4.21mg、芍药新苷甲素对照品2.15mg,置10mL量瓶中,加甲醇溶解并稀释至刻度,即得对照品溶液;
(3)供试品溶液的制备:精密量取药物样品0.5g,精密加入甲醇25mL,称重,超声提取30min,取出,放冷,补重,摇匀,0.45μm微孔滤膜滤过,取续滤液,即得供试品溶液;
(4)测定:精密吸取对照品溶液和供试品溶液各10μL注入高效液相色谱仪,进行测定。
2、采用高效液相色谱法测定药物中消旋-丁香树脂酚的含量:
(1)色谱条件:采用HypersilDs色谱柱;规格:4.0mm×125mm,5μm,流动相:比例为80:20的乙腈-0.05%磷酸溶液;检测波长:268nm;柱温:35℃;流速:1.0mL·min-1;
(2)对照品溶液的制备:精密称取80℃干燥至恒重的消旋-丁香树脂酚对照品适量,加甲醇制成每1mL含0.2mg的溶液,即得对照品溶液;
(3)供试品溶液制备:精密称取药物样品10g,加甲醇40mL,加热回流4h,提取液回流溶剂并浓缩至干,残渣加水10mL溶解,用水饱和的正丁醇振摇提取5次,每次20mL,合并正丁醇提取液,用氨试液洗涤3次,每次15mL,正丁醇提取液回收溶剂至干,残渣加甲醇溶解并转移至10mL容量瓶中,加甲醇至刻度,摇匀,滤过,取滤液,即得供试品溶液;
(4)测定:精密称取对照品溶液和供试品溶液各10μL,注入高效液相色谱仪进行测定;
(5)测定结果:药物样品中消旋-丁香树脂酚的含量为0.185mg·g-1。
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的技术方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
- 还没有人留言评论。精彩留言会获得点赞!