基于生物信息学分析植物性食物过敏原线性表位的方法与流程
本发明涉及食品安全检测技术领域,尤其涉及一种基于生物信息学分析植物性食物过敏原线性表位的方法。
背景技术:
食物过敏是机体暴露于某种特定食物时出现的不良反应,即免疫系统对过敏原进行特异性免疫应答所致的可涉及全身不同器官的过敏反应。据流行病学调查全世界范围内婴幼儿的发病率已达到8%,成人达到5%。食物过敏大多是由ige介导的i型超敏反应,可引发哮喘、腹泻、荨麻疹、过敏性皮炎等全身性多系统反应,甚至会产生休克危及生命,严重影响患者的生活质量。迄今为止,防止食物过敏最有效的途径是避免接触食物过敏原。但由于生产过程中的交叉反应增加了食品存在过敏原的可能性,食物过敏患者必须倍加小心才能免受其害。
自然界中的食物过敏原种类繁多,主要来源于植物、动物和微生物,但是引起食物过敏性疾病的过敏原大多数来源于植物。到目前为止,世界卫生组织/国际免疫学会联合会(who/iuis)过敏原命名小组委员会共鉴定了314种食物过敏原,其中有216种来源于植物。而联合国粮农组织(fao)报道的八类过敏性食物中有四类是植物性食物,即花生、大豆、坚果类和小麦。
食物过敏原分子表面具有特定的结构和氨基酸序列,可以刺激机体产生免疫反应,该免疫活性区被称为抗原表位。根据抗原表位中氨基酸的空间分布特点,可以分为连续性表位和不连续性表位。连续性表位又称为线性表位,是由一级序列中氨基酸连续组成;不连续性表位又称为构象表位,在一级序列中并不连续,但能在正确折叠的蛋白空间结构表面彼此靠近,形成被抗体特异性识别的表位。研究表明,在所有抗原表位中超过90%属于构象表位,只有不到10%是线性表位。但食物过敏原经口摄入后会被胃和小肠消化,胃酸、消化酶等因素均会破坏大量的构象表位,而线性表位依然能被鉴定出,并刺激机体产生过敏反应。因此,食物过敏原线性表位的研究对预防和治疗食物过敏至关重要。
同时,植物性食物过敏原蛋白质在三维结构等方面具有保守相似性,植物性食物过敏原的高度稳定性与其紧凑的三维结构以及维系空间结构的化学键密不可分。因此,对植物性食物过敏原高级结构的研究及其与线性表位关系的研究,有助于分析蛋白质的潜在致敏性。
目前,用于鉴定分析线性表位的方法主要有多肽合成技术和氨基酸定点突变技术等。多肽合成技术是将合成的短重叠肽段通过斑点印迹或免疫印迹实验与过敏患者血清共孵育,检测其与特异性ige抗体的结合情况,从而判断和筛选线性表位,但需要收集过敏患者血清,耗时长、工作量较大、盲目性较高,有可能忽略重叠区间的表位,难以保证精准性。氨基酸定点突变技术是通过依次突变目的蛋白的某个或几个特定氨基酸,然后比较天然目的蛋白和突变后重组蛋白与抗体的结合程度筛选出主要的表位,整个过程突变、重组和筛选工作量都很大。
生物信息学是通过对生物信息的获取、处理、分析,来阐明大量数据所包含的生物学意义的一门新兴交叉学科。本发明旨在通过生物信息学方法深入研究线性表位,无需实验,快速准确。
技术实现要素:
针对现有技术存在的问题,本发明提供一种基于生物信息学分析植物性食物过敏原线性表位的方法。
本发明提供一种基于生物信息学分析植物性食物过敏原线性表位的方法,包括以下步骤:
(1)利用生物信息学数据库获取植物性食物过敏原的线性表位、氨基酸序列、二级结构和三级结构;
(2)利用生物信息学软件分析所述线性表位,包括其氨基酸组成及出现频率,在所述二级结构中的位置和在所述三级结构中的位置。
上述技术方案中,利用生物信息学数据库及软件获取和分析植物性食物过敏原的氨基酸序列、二级结构和三级结构等,从而分析植物性食物过敏原线性表位与其高级结构的位置关系及特性,既快速准确又无需实验,对线性表位的深入研究有助于对食物致敏性的研究。
所述生物信息学数据库包括但不限于sdap、pdb、uniprot、ncbi和blast。
所述生物信息学软件包括但不限于bioedit、cn3d、dssp、swiss-model、spdbv、pymol、clustalw、pfind和dnastar。
进一步地,所述线性表位不能直接获取时,通过以下方法预测获得:利用生物信息学软件分别筛选出所述植物性食物过敏原氨基酸序列中满足亲水性指数>0的区域、抗原指数>0的区域和表面可及性指数>1的区域,并利用可塑性分析筛选出具有柔性的区域,上述四个区域重叠的部分即为预测线性表位。
进一步地,所述三级结构不能直接获取时,通过同源建模预测获得。
进一步地,所述同源建模预测具体包括:在pdb数据库中使用blast搜索与所述植物性食物过敏原同源性较高,且已知三级结构数据的蛋白质序列;以相似度最高的蛋白质为模板,使用swiss-model在线网站进行相同结构区域的同源建模,根据分子动力学模拟和能量最小化原理优化得到所述植物性食物过敏原的三级结构。
进一步地,所述方法还包括:使用spdbv软件获得所述植物性食物过敏原三级结构的拉氏构象图以评价其合理性。
进一步地,所述二级结构不能直接获取时,依据所述植物性食物过敏原的氨基酸序列,使用dnastar软件中的chou-fasman或garnier-robson方案进行预测。
进一步地,所述方法还包括利用生物信息学软件对所述植物性食物过敏原进行同源性分析。
进一步地,利用cn3d或pymol软件分析所述线性表位在所述三级结构中的位置。
本发明利用生物信息学数据库及软件分析植物性食物过敏原的氨基酸序列、二级结构和三级结构等,从而分析植物性食物过敏原线性表位与其高级结构的位置关系及特性,既快速准确又无需实验,对线性表位的深入研究有助于对食物致敏性的研究。
附图说明
为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
图1为本发明实施例中arah1全序列的氨基酸组成情况;
图2为本发明实施例中arah1线性表位的氨基酸组成情况;
图3为本发明实施例中arah1的二级结构分布情况;
图4为本发明实施例中arah1三聚体(左)和单体(右)结构;
图5为本发明实施例中arah1线性表位在三级结构中的定位;
图6为本发明实施例中jugr2的同源性模板检索结果;
图7为本发明实施例中jugr2的模拟三级结构;
图8为本发明实施例中jugr2模拟三级结构的拉氏构象图;
图9为本发明实施例中jugr2与arah1的序列比对结果;
图10为本发明实施例中jugr2与arah1相似肽段在三级结构中的定位;
图11为本发明实施例中dnastar对cm16一级氨基酸序列的分析;
图12为本发明实施例中cm16的二级结构预测;
图13为本发明实施例中cm16的同源性模板检索结果;
图14为本发明实施例中cm16的模拟三级结构;
图15为本发明实施例中cm16模拟三级结构的拉氏构象图;
图16为本发明实施例中cm16预测线性表位在其三级结构中的定位。
具体实施方式
为使本发明实施例的目的、技术方案和优点更加清楚,下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
实施例1
花生过敏原arah1为7s球蛋白,属于cupin超家族,经血清学分析发现可被90%以上的花生过敏患者血清识别。由于cupin超家族的结构序列具有高度保守性、热稳定性强、抗酶解和不易消化等特点,所以arah1被选为本发明中的模式过敏原。
本实施例提供一种基于生物信息学分析花生过敏原arah1线性表位的方法,包括以下步骤:
(1)在uniprot数据库获取arah1的氨基酸全序列;在致敏蛋白结构数据库sdap中获取arah1的23个线性表位序列,如表1所示。
表1花生主要过敏原arah1的线性表位序列
注:下划线标注为ige结合多肽的关键氨基酸残基。
(2)利用bioedit软件对arah1全序列及线性表位的氨基酸组成和出现频率进行分析
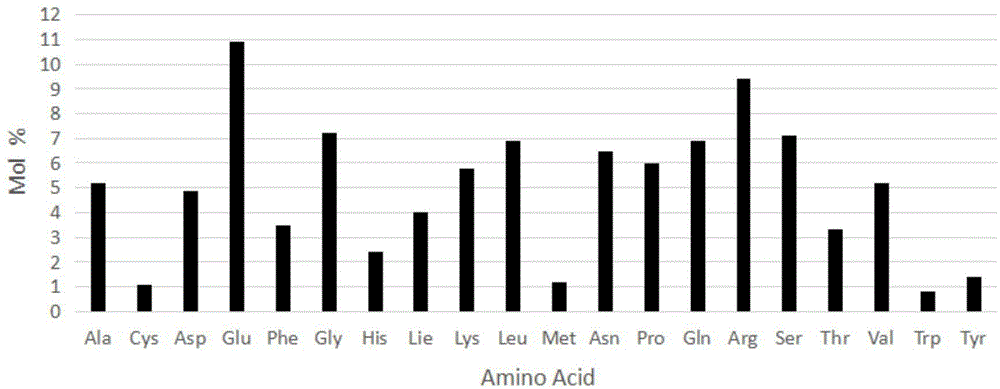
使用bioedit软件分析arah1一级序列的氨基酸组成及出现频率,如图1所示,发现arah1全序列由20种氨基酸组成,含量最多的为谷氨酸(glu)和精氨酸(arg)。同样使用该软件分析了arah1线性表位的氨基酸组成及出现频率,如图2所示,arah1的线性表位由19种氨基酸组成,其中天冬氨酸(asp)、苯丙氨酸(phe)、甘氨酸(gly)、脯氨酸(pro)和精氨酸(arg)的出现频率明显升高,证明其为关键活性氨基酸。
(3)利用dssp软件对arah1二级结构与线性表位的分析
从pdb数据库中获取了arah1的三级结构,使用dssp二级结构分析程序计算出二级结构的分布情况,如图3所示。从图中可以看出arah1的二级结构主要是由α-螺旋束和β-折叠组成的,全长包含418个氨基酸,对应于arah1一级序列的170-587位,涵盖了10-22号线性表位。线性表位大多位于α-螺旋与无规则卷曲、β-折叠与β-转角的相交处。
(4)利用cn3d软件对arah1线性表位在三级结构中定位的分析
从pdb数据库可知天然状态下arah1可以由3个同源亚基连接成稳定的三聚体结构,如图4所示,组成三聚体的arah1单体则可以分为4个结构域,两个α-螺旋束位于亚基的两端,中间是两个相互对称的反向平行β-折叠结构域。使用cn3d软件将arah1的10-22号线性表位标注于其中一个单体上,如图5所示,其中深棕色单体上的黄色区域即为标注的线性表位,从图中可以看出,大部分线性表位都位于单体之间的疏水相互作用结合区域,该区域有利于保护线性表位,使其难以被蛋白酶消化,该结果与线性表位的氨基酸组成分析具有一致性,即疏水氨基酸的含量显著高于在全序列中的分布。
综上可知,cupin超家族线性表位的中央部分多由疏水性氨基酸如苯丙氨酸(phe)和脯氨酸(pro)组成,并富含天冬氨酸(asp)和精氨酸(arg)等带电氨基酸;在二级结构中,线性表位多位于两种结构相连的变化区域,具有一定的回转折叠构象;在三级结构中,线性表位主要位于单体之间的疏水相互作用区域,多埋入三聚体构象内部。
实施例2
核桃过敏原jugr2属于7s豌豆球蛋白家族,与花生过敏原arah1同家族,一级序列由593个氨基酸组成,分子量约为44kda。
本实施例提供一种基于生物信息学分析核桃过敏原jugr2线性表位的方法,包括以下步骤:
(1)jugr2三级结构的同源建模预测
在pdb数据库中使用blast搜索与过敏原jugr2同源性较高,且已由实验分析出三级结构数据的蛋白质序列,结果如图6所示,其中pdb检索号为5e1r.1.a的7s豌豆球蛋白在序列相似性和覆盖率方面匹配度最高,选为jugr2建模的最佳模板。5e1r.1.a是山核桃的7s豌豆球蛋白食物过敏原的晶体结构,全长426个氨基酸,与jugr2的序列相似性可达90.69%。
使用swiss-model在线网站构建jugr2的三级结构,得到的拓扑卡通图如图7所示,根据二级结构的组成着色,蓝色标注为分子内α-螺旋所在的位置,绿色标注为β-折叠。由图可知,jugr2的三级结构是由3个亚基首尾相连形成的同源三聚体,每个亚基分子结构中包含位于中间的2个反向平行cupin结构域,以及在两端的α-螺旋束,呈现出典型的cupin超家族结构。
使用spdbv软件获得模拟三级结构的拉氏构象图,从而进行合理性分析。拉氏构象图是α-碳与酰胺平面的交角图,可以用来评价蛋白质结构中转角的易变程度。通过了解氨基酸残基的phi和psi角信息,获得拉氏构象图的允许构象和不允许构象区域,如图8所示,其中在空间上允许和不允许出现的区域用不同颜色的等高线表示,黄色封闭区域为允许区,构象最为稳定,蓝色封闭区域为临界区,不够稳定,除此之外的区域为不允许区,图中红色的点为组成α-螺旋的氨基酸,黄色的为β-折叠。由图可知只有8个氨基酸位于不允许区,大部分的氨基酸都分布在允许区内,此结果表明jugr2的建模成功,其三级结构具有较高的合理性和稳定性。
(2)jugr2序列同源性分析
从pdb数据库中获取花生主要过敏原arah1的氨基酸序列、二级结构和三级结构,从ncbi数据库中获取核桃主要过敏原jugr2的氨基酸序列;使用ebi中的clustalw在线分析软件对arah1和jugr2进行序列比对,分析其保守和非保守区域;用espript3.0软件在比对的序列中呈现相似性和二级结构。
将jugr2与arah1进行序列比对的结果如图9所示,从图中可以看出,jugr2与arah1具有一定的序列相似度(34.34%),且保守区域大部分存在于二级结构形成区域,保证了cupin超家族蛋白质在结构上的高度相似性。同时也说明了cupin超家族过敏原具有保守相似性,可以利用模式抗原arah1进行代表性研究。
(3)jugr2与arah1相似肽段的空间定位
利用pymol软件对jugr2与arah1相似肽段的序列在三维结构中进行定位。如图10所示,将之前同源对比获取的多个肽段按不同颜色标注在jugr2的空间结构中。图a显示的是jugr2的拓扑卡通图,彩色区域是不同肽段在其中的定位,由图可知,除了标记为绿色的215-220位点位于三聚体的核心区域外,其余肽段均位于空间结构的表面或亚基之间的疏水作用结合区域,其二级结构多为β-转角或无规则卷曲等暴露于分子表面的可变性区域,具有良好的抗体结合构象,有利于线性表位的形成。
图b1和b2显示的是jugr2分子的表面暴露情况,具体分别是蛋白质分子的正视图和亚基连接部位的侧视图,肽段标记的颜色与图a相同。由图可知,绿色的215-220区域位于结构的核心处,不具有溶剂可及性和接触面积,而其余肽段均有部分序列暴露在外,有利于在溶液环境中与抗体产生相互作用,可能含有线性表位。
综上可知,利用swiss-model对jugr2的三级结构进行同源建模,选用山核桃7s过敏原5e1r.1.a为模板,其与jugr2序列相似性为90.69%,发现jugr2具有典型的cupin超家族蛋白构象,使用拉氏构象图对建模结果进行评估,确定模型的构象合理性。对arah1和jugr2的序列相似性进行比对,结果表明,这两种蛋白在构成二级结构的氨基酸组成上具有高度相似性,用以维持cupin超家族独特的构象。同时,jugr2中7条肽段(下划线所示)与arah1的线性表位(方框所示)存在重合位点,表明jugr2很有可能会引起arah1过敏患者产生交叉过敏反应。jugr2具有与arah1线性表位相同的分布规律,证明本发明建立的生物信息学分析植物性食物过敏原线性表位的方法对相同家族植物过敏原具有适用性。
实施例3
小麦过敏原cm16是分子量为17kda,由143个氨基酸组成的蛋白质,属于醇溶蛋白超家族,含有多个半胱氨酸残基,可构成分子内二硫键,保证蛋白质的结构稳定性,并具有耐热性。
本实施例提供一种基于生物信息学分析小麦过敏原cm16线性表位的方法,包括以下步骤:
(1)cm16线性表位的预测
从uniprot数据库获取小麦过敏原cm16的氨基酸序列,使用生物信息学软件dnastarprotean中的hoop-woods的氨基酸亲水性分析方案、emini-surfaceprobability的溶剂可及性算法、kparlus-schuzl的可塑性分析和jameson-wolf的抗原指数分析方案,对cm16可能的线性表位进行分析。
结果如图11所示,在氨基酸亲水性分析中,亲水性指数>0表明亲水性好,cm16的亲水性区域分布较为均匀,其中亲水性较高的区域分别为aa88-100和aa137-143;在可塑性分析中,发现aa26-32、aa39-51、aa54-62、aa64-67、aa75-78、aa89-98、aa103-108、aa130-132和aa138-140区域具有一定的柔性,容易发生折叠、弯曲,易与抗体结合;在抗原指数分析中,以抗原指数>0为筛选条件,结果显示抗原指数较高的区域为aa26-34、aa42-63、aa66-70、aa71-84、aa88-99、aa100-111和aa137-143,可能含有潜在的优势抗原表位;在表面可及性分析中,以表面可及性指数>1为筛选条件,结果显示aa47-50、aa58-60、aa62-65、aa90-96和aa138-143区域具有较好的表面可及性。
综合以上各参数,将同时满足4个参数筛选条件的表位预测为cm16的线性表位,具体如表2所示。
表2dnastar预测得到的cm16的线性表位
(2)cm16二级结构的预测
使用dnastarprotean载入ncbi中下载的fasta格式的cm16氨基酸序列,使用chou-fasman和garnier-robson方案分别对蛋白质的二级结构进行预测分析,结果如图12所示。从图中可以看出,两种方案在预测cm16的β转角时分析结果具有一定的相似性。总的来看,cm16的二级结构中有序的螺旋、转角结构占据了主要优势,反映了蛋白质具有良好的紧密结构。
(3)cm16三级结构的同源建模预测
在pdb数据库中使用blast搜索与过敏原cm16同源性较高,且已由实验分析出三级结构数据的蛋白质序列,结果如图13所示,其中pdb检索号为1bfa.1.a的淀粉酶抑制剂与cm16的序列相似性可达41.88%,具有较高的序列覆盖率,模拟结构评分最高,因而被选为cm16建模的最佳模板。
使用swiss-model在线服务器构建cm16的三级结构,得到的拓扑卡通图如图14所示,根据二级结构的组成着色,蓝色标注为分子内α-螺旋所在的位置。由图可知,cm16的三级结构是由4个分子内的α-螺旋以及其他无序结构组成的,具有醇溶蛋白超家族典型的结构特性。
使用spdbv软件获得模拟三级结构的拉氏构象图,对cm16进行构象合理性分析,结果如图15所示,其中红色的点代表组成α-螺旋的氨基酸。由图可知,红色点主要分布在第三象限的允许区域内,而其他颜色的点主要分布在第二象限的允许区域内,由此可证,cm16的二级结构主要由α-螺旋组成;只有6个点位于不允许区域,允许区域的氨基酸覆盖率达88%,此结果表明构建的cm16的三级结构具有合理性和稳定性。
(4)cm16预测线性表位的空间定位
利用pymol软件对预测得到的cm16线性表位在其三级结构上进行定位,结果如图16所示,图中将模拟的4条线性表位按不同颜色标注在三级结构图上。图a为卡通拓扑图,除了标注为红色的线性表位位于α-螺旋上以外,其他的三个线性表位均位于无规则卷曲处,构象上具有可塑性,容易与抗体接触形成表位。图b显示的是cm16的表面分子暴露情况,可以看出4个预测的线性表位均位于球状结构的表面,具有与抗体结合的表面可及性和暴露面积,可以形成线性表位。
综上可知,cm16的4条可能的线性表位都位于氨基酸序列的中部和c末端,cm16是由4个α-螺旋以及无规则卷曲组成的球状结构,预测的线性表位位于无规则卷曲处或是α-螺旋与无规则卷曲的连接处,且暴露于球状结构的表面,具有溶剂可及性及结构可塑性。预测表位的空间定位具有表面可及性和结构可塑性等分布规律,证明本发明建立的生物信息学分析植物性食物过敏原线性表位的方法同样适用于醇溶蛋白超家族。
综合以上结果,本发明建立了利用生物信息学数据库及软件分析植物性食物过敏原线性表位的方法,适用于所有植物性食物过敏原,此方法准确、快速且无需实验。
最后应说明的是:以上实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的精神和范围。
- 还没有人留言评论。精彩留言会获得点赞!