一种低密度SNP基因组区域准确预测BSA-seq候选基因的方法与流程
本发明涉及基因组测序技术领域,特别涉及一种低密度snp基因组区域准确预测bsa-seq候选基因的方法。
背景技术:
集团分离分析法(bulkedsegregantanalysis,bsa)是1991年由r.w.michelmore在莴苣上首次应用的一种快速定位控制目标性状基因的方法。方法是取子代群体中具有极端表型的单株,等量pooling其dna形成两个dna池,然后在亲本和两个池之间进行标记多态筛选,通过对子代群体筛选得到的多态标记进行基因型分析,即可完成对目标基因的定位,而不需要对每个标记都在群体里进行基因型分析。随着高通测序技术的兴起,基于全基因组重测序的bsa分析方法广泛应用在植物重要性状定位中,其具有“快速、高效、价廉”等特点。bsa-seq的基本思路,通常是指从作图群体中挑选极端个体,然后等量混合样本构成两个dna池,对亲本和池进行高通量测序,鉴定在亲本和两个池中共有的snps,计算两个混合dna池中相同变异位点的基因型频率及其差值,以差值来体现标记在池间的多态性,从而实现候选基因的定位然而,bsa-seq相对于全基因组关联分析、遗传图谱等基因定位技术,存在准确度低、精确度低等缺点,如何对低密度snp区域的候选基因进行准确预测,是我们面临的难题,更多时候在基因组差异小的区域容易造成的候选区域的假阳性。
技术实现要素:
鉴于上述内容,有必要针对差异小的基因组区域对候选基因进行准确预测,并提供一种快速、高效、廉价的预测方法。
为达到上述目的,本发明所采用的技术方案是:
一种低密度snp基因组区域准确预测bsa-seq候选基因的方法,所述方法包括如下步骤:
(1)混池:选择目标性状差异显著的亲本构建分离群体,然后从分离群体中选择极端表型的若干个单株分别混合成两个等量的dna池;
(2)提取dna:提取植物基因组dna;
(3)测序:检测步骤(2)的dna样品,合格后将dna片段化,对dna片段进行修饰、pcr扩增,构建测序文库,文库质检合格后进行测序;
(4)比对:将步骤(3)获得的测序reads重新定位到参考基因组上,进行比对、统计,计算相对于参考基因组的测序深度和覆盖度;
(5)snp检测与注释:使用gatk软件进行snp的检测;利用软件snpeff进行注释变异和预测变异;
(6)snp-index关联分析:对snp进行过滤,进行频率差异分析、计算得到snp-index及△snp-index的分布;
(7)候选区间分析:根据步骤(6)△snp-index的分布情况,选择低密度区域为候选区间,利用基因组注释网站对候选区间基因进行注释;对候选区间变异位点进行功能注释,找出存在移码变异等功能性变异的基因,得到候选基因;利用qrt-pcr技术对候选基因进行验证。
进一步的,其特征在于,所述步骤(6)snp过滤的过滤标准如下:首先,过滤掉有多个基因型的snp位点,其次,过滤掉reads支持度小于4的snp位点,再次过滤掉混池之间基因型一致的snp位点以及隐性混池基因不是来自于隐性亲本的snp位点。
进一步的,所述步骤(6)的计算采用snp-index方法计算关联值,并采用distance方法对△snp-index进行拟合。
进一步的,所述所述步骤(5)snp检测与注释方法如下:
步骤s1:通过gatk软件工具包检测测序基因组的snp和smallindel;通过bwa软件,采取mem算法将高质量的测序reads比对到参考基因组,根据cleanreads在参考基因组的定位结果,使用picard过滤冗余reads;使用gatk的局部单体型组装算法进行snp和indel的变异检测,每个样本先各自生成gvcf,再进行群体joint-genotype得到变异位点集;并对变异结果进行过滤得到过滤后的snp列表,所述过滤标准为:5bp窗口内的变异数量不超过2个;phred格式的质量值不低于30;变异质量值除以覆盖深度的比值不低于2.0;所有比对至该位点上的reads的比对质量值的均方根不低于40;fs值不高于60;其它变异过滤参数采用gatk官方指定的默认值处理。
步骤s2:基于步骤s1得到过滤后的snp列表,通过定制化的脚本获得在父本池和母本池具有差异的位点即是亲本之间的snp位点,然后通过滑窗统计snp的分布密度,定制化的脚本画分布图。
本发明的另一目的还包括上述方法在植物基因标记中的应用。
进一步的,所述植物为水稻。
进一步的,所述水稻的亲本为黄华占和东兰墨米。
进一步的,所述植物基因为水稻种皮花青素合成基因。
进一步的,所述花青素合成基因为loc_os01g44260。
本发明具有如下有益效果:
本发明针对bsa-seq在候选区间附近有低密度snp区域,通过比价两亲本间的snp,对snp列表进行严格过滤,找出低密度区域,然后利用置信区间为95%时对应的候选区间加上低密度候选区间,利用基因组注释网站对候选区域内的基因进行注释;对候选区域变异位点功能注释,得到存在移码变异等功能性变异的基因,并确定该基因为候选基因;使用本发明的方法能弥补由于基因组差异小的区域而造成的候选区域的假阳性,获得真正的候选区间。
【附图说明】
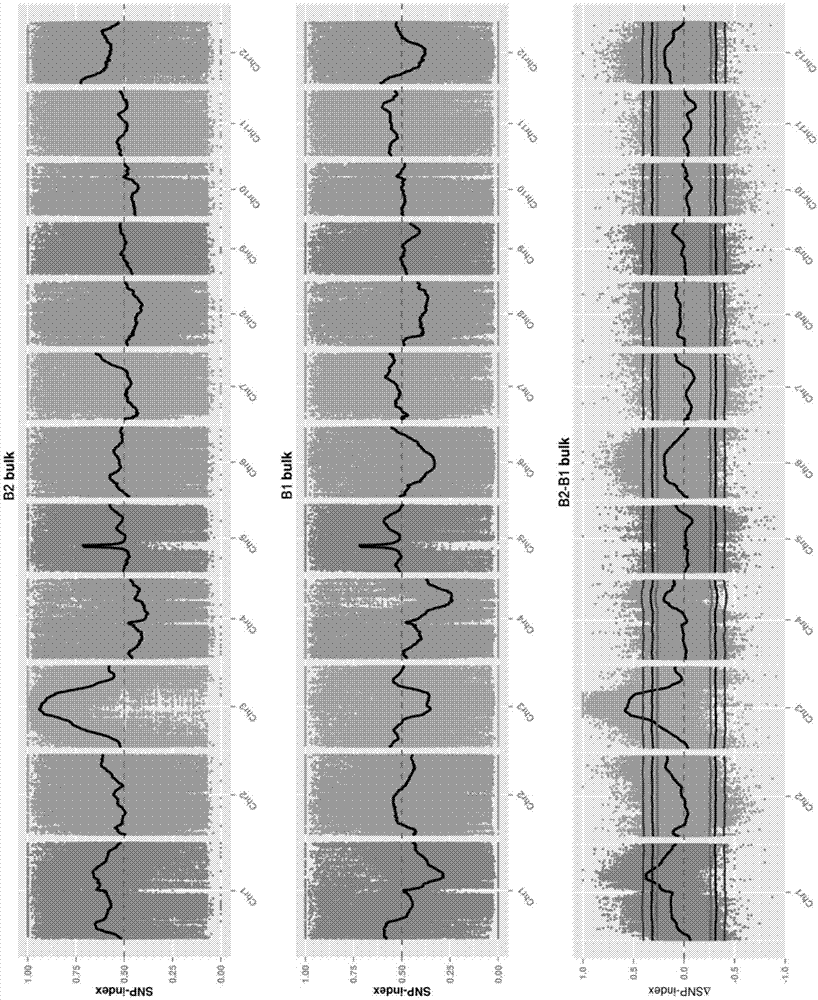
图1是本发明实施例候选基因置信区间的分析图;
图2是本发明实施例基因组上的snp分布图。
【具体实施方式】
本说明书中公开的所有特征,或公开的所有方法或过程中的步骤,除了互相排斥的特征和/或步骤以外,均可以以任何方式组合。
本说明书(包括任何附加权利要求、摘要)中公开的任一特征,除非特别叙述,每个特征只是一系列等效或类似特征中的一个例子而已。
实施例:
本实施例提供了一种低密度snp基因组区域准确预测bsa-seq候选基因的方法,该方法步骤如下:
(1)混池:选择目标性状差异显著的亲本“黄华占×东兰墨米”构建分离群体,再从分离群体中选取目标性状表型极端的30-50个单株,分别混合成两个dna池(dnapools)进行测序;
(2)提取dna:采用ctab法提取植物基因组dna;
(3)测序:步骤(2)得到的基因组dna样品检测合格后,用超声破碎的方法将dna随机打断成350bp的片段,对dna片段进行修饰,修饰方法为:对dna片段进行末端修复、磷酸化并加ploy(a)、加测序接头;然后进行纯化、pcr扩增,构建测序文库;文库经质检合格后通过illuminahiseqxten进行测序,并对获得的reads进行质量控制;
(4)与参考基因组比对统计:对步骤(3)重测序获得的reads重新定位到参考基因组上。bwa软件主要用于二代高通量测序得到的短序列与参考基因组的比对。通过比对定位cleanreads在参考基因组上的位置,统计各样品的测序深度、基因组覆盖度等信息,并进行变异的检测;
(5)snp检测与注释:snp的检测主要使用gatk软件实现;注释变异(snp、smallindel)和预测变异影响利用软件snpeff进行。
(6)snp-index关联分析:首先对snp进行过滤,过滤标准如下:首先过滤掉有多个基因型的snp位点,其次过滤掉reads支持度小于4的snp位点,再次过滤掉混池之间基因型一致的snp位点以及隐性混池基因不是来自于隐性亲本的snp位点;利用snp-index方法计算关联值,并采用distance方法对△snp-index进行拟合。两个混池分别的snp-index及△snp-index的分布。
(7)候选区间分析:如图1所示,选择95%置信区间对应的阈值以上的基因组区,该基因组区位于水稻的第1染色体上26.57mb-31.55mb区间内;我们在这段区域利用水稻基因组注释网站msu-rgap预测基因注释、候选区域变异位点功能注释、候选基因表达分析等但并未找到候选基因;经进一步分析,如图2所示,在26.57mb-31.55mb区间的上游19.73-26.50mb区域内snp数量突然下降,这可能是造成bsa-seq候选区域不准确的重要原因,因此,我们选择其上游19.73-26.50mb区域为候选区,利用水稻基因组注释网站msu-rgap预测基因注释、候选区域变异位点功能注释、候选基因表达分析等,获得水稻种皮花色素合成基因的候选基因loc_os01g44260。
本实施例还对snp检测与注释主要进行了如下质量控制:
步骤s1:基于水稻参考基因组获取snp和indel,具体方法如下:snp(singlenucleotidepolymorphism,单核苷酸多态性)和smallindel(smallinsertionanddeletion,小片段的插入与缺失)的检测主要使用gatk软件工具包实现。通过bwa软件,采取mem算法将高质量的测序reads比对到水稻参考基因组,根据cleanreads在参考基因组的定位结果,使用picard过滤冗余reads(markduplicates),以保证检测结果的准确性。然后使用gatk的haplotypecaller(局部单体型组装)算法进行snp和indel的变异检测,每个样本先各自生成gvcf,再进行群体joint-genotype得到变异位点集。为了保证变异结果的可靠性,变异结果经过严格的过滤,主要过滤参数如下:
①5bp窗口内的变异数量不应该超过2个;
②qual<30,(qual为:phred格式的质量值,表示该位点存在variant变异的可能性)。
质量值低于30的则过滤掉;
③qd<2.0,(qd为:变异质量值除以覆盖深度得到的比值,覆盖深度是这个位点上所有含有变异碱基的样本的覆盖深度之和)。qd低于2.0的则过滤掉;
④mq<40,(mq为:所有比对至该位点上的reads的比对质量值的均方根)。mq低于40的则过滤掉;
⑤fs>60,(fs为:通过fisher检验的p-value转换而来的值,描述的是测序或者比对时对于只含有变异的reads以及只含有参考序列碱基的reads是否存在着明显的正负链特异性)。也就是说,不会出现链特异的比对结果,fs应该接近于零。fs高于60的则过滤掉;
⑥其它变异过滤参数采用gatk官方指定的默认值处理。
步骤s2:基于步骤s1得到的严格过滤的snp列表,通过定制化的脚本获得在父本池和母本池具有差异的位点即是亲本之间的snp位点,然后通过滑窗统计snp的分布密度,通过定制化的脚本画分布图。
综上所述,使用本申请的方法,能针对差异小的基因组区域对候选基因进行准确预测,而且本发明的方法还具有快速、高效、廉价的优势。
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明的保护范围应以所附权利要求为准。
- 还没有人留言评论。精彩留言会获得点赞!