一种基于排挤策略的多模态蛋白质结构预测方法与流程
本发明涉及一种生物信息学、智能优化、计算机应用领域,尤其涉及的是一种基于排挤策略的多模态蛋白质结构预测方法。
背景技术:
science于2005年发表的125个科学问题中提出“蛋白折叠能否被预测的问题”,表明以蛋白质为代表的生物大分子结构测定,不仅是分子生物学中心法则尚未解决的基础理论问题,更是关乎人类切身利益的医学、药学及材料学等领域亟待解决的应用问题。比如,朊蛋白(prionprotein,prp)的结构变异将引发疯牛病,而蛋白质错误折叠会导致阿兹海默症、帕金森氏症等疾病。因此,深入了解蛋白质天然态结构与其折叠机理将有助于阐明这些疾病的致病机制并寻找治疗方法。
结构生物学实验是测定生物大分子结构的主要技术手段。卡文迪许实验室于60年代用x-射线晶体衍射技术获得了球蛋白的结构,从晶体水平进行生物大分子的研究;多维核磁共振(nmr)方法可以直接测定蛋白质在溶液中的构象,更接近于生理状态;近期发展的冷冻电镜技术,通过电子显微镜技术并结合图像处理技术可直接提供生物大分子的形貌信息。目前pdb中的蛋白质三维结构主要通过结构实验测定技术得到,不仅存在应用局限,测定过程也费时费钱费力,比如,使用nmr方法测定一个蛋白质通常需要15万美元以及半年的时间,因此实验测定蛋白质三维结构的速度远远达不到所需速度。鉴于结构测定耗资巨大以及信息科学技术的飞速发展,以计算机为工具,运用适当的算法,从序列出发直接预测生物大分子的三维结构,在后基因组时代生命科学的理论和应用研究中将发挥日益重要的作用。
不依赖于任何已知结构,基于anfinsen热力学假说,构建蛋白质能量函数,通过有效的构象搜索方法来搜寻目标蛋白的天然结构。anfinsen热力学假说认为蛋白质的天然结构具有热稳定性,对应于蛋白质及周围溶剂分子所构成的整个体系的自由能极小点,因此从头预测方法通常采用片段组装技术,在构象搜索过程中指导算法向低能量构象进行搜索。通过全局优化方法确定蛋白质的结构,是基于其全局极小描述蛋白质天然结构的打分函数,然而分子内作用与周围环境作用的复杂性,将形成一个具有大量局部极小的粗糙能量地貌,由于能量模型的复杂性和不精确性,使得数学上的最优解并不一定对应其稳定的天然结构,有时局部极值解才真正与蛋白质实测基态构型吻合。由于能量模型的复杂性和不精确性,使得数学上的最优解并不一定对应其稳定的天然结构,有时局部极值解才真正与蛋白质实测基态构型吻合。另外,鉴于粗糙不平的表面是蛋白质能量地貌的固有属性,计算量将随着分子的大小呈指数增长。基于上述考虑,有必要从蛋白质结构预测方法的多模态特性方面进行深入研究。
因此,目前的蛋白质结构预测方法在对能量模型多极值解的搜索上存在不足,需要改进。
技术实现要素:
为了克服现有的蛋白质结构预测方法在搜索能量模型多极值解上的不足,本发明提出一种基于排挤策略的多模态蛋白质结构预测方法,该方法在差分进化算法的框架下,采用排挤策略,在进化过程中自适应地形成多个模态,使之能够发现模型所有的局优解,并且在此过程中尽可能多地保存局优解,从而提高蛋白质结构预测方法的预测精度。
本发明解决其技术问题所采用的技术方案是:
一种基于排挤策略的多模态蛋白质结构预测方法,所述方法包括以下步骤:
1)给定输入序列信息,以及蛋白质力场模型,即能量函数rosettascore3;
2)初始化:迭代rosetta协议第一、二阶段,产生具有np个构象的种群pg,记为
3)通过差分进化算法的交叉、变异操作生成种群pg的试验构象种群ug,记为
3.1)从种群pg中选定目标个体
3.2)在[0,l-9]内生成均匀随机整数rand1、rand2和rand3,其中l表示氨基酸序列长度;
3.3)将
3.4)i=i+1,循环步骤3.1)-3.4)直至生成当前种群的试验个体种群ug;
4)为种群pg中的每一个构象
4.1)对ug中的每一个试验构象
4.2)t=t+1,重复步骤4.1),直至t=np,记此时
4.3)将
4.4)i=i+1,循环步骤4.1)-4.4)直至为每一个构象
5)对每一个
其中
6)排挤操作:用
7)聚类操作:生成当前第g+1代种群的模态构象集合
7.1)mg+1初始化为只有一个构象的集合,该构象为种群pg+1中的最好构象;
7.2)将
其中
7.3)i=i+1,循环步骤7.1)-7.3)直至为第g+1代种群找到所有的模态构象;
8)判断是否满足终止条件,若满足则输出结果并退出,否则g=g+1清空mg+1并且返回步骤3)。
进一步,所述步骤2)中,设置最大迭代次数gmax,所述步骤8)中,对种群执行完步骤3)-7)以后,终止条件为迭代次数g达到预设最大迭代次数gmax,若满足输出结果为
本发明的技术构思为:首先,对初始种群中的每个构象构建各自的存档集合,用以获得对应的聚类中心和聚类半径,并且根据存档集合进行排挤操作以更新种群;其次,通过比较当前种群构象的聚类半径,对每一代种群构建当前的模态集合,即确定了当前代的模态构象个数k;最后,随着迭代的进行,模态构象集合逐渐稳定,得到k个局优构象,最终得到全局最优构象。
本发明的有益效果表现在:由于能量模型的不精确性,导致其全局和局部最优均有可能是最好构象。在差分进化算法的框架下,通过排挤操作将种群自动聚类,形成多个模态,一方面,模态构象数随着种群进化情况而自适应变化,直至稳定,从而能够搜索到多个局优构象以缓解能量模型的不精确问题;另一方面,该排挤策略只对差分进化算法中的选择环节进行了修改,并未增加算法复杂度,操作简单可行。
附图说明
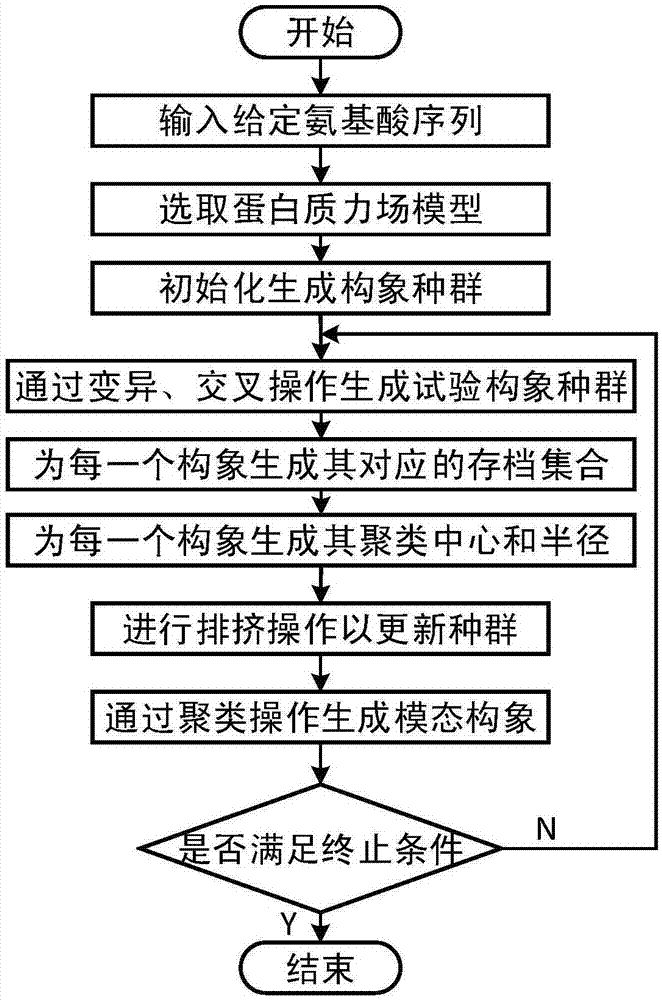
图1是基于排挤策略的多模态蛋白质结构预测方法对蛋白质1hz6进行结构预测时的流程图;
图2是基于排挤策略的多模态蛋白质结构预测方法对蛋白质1hz6进行结构预测时的构象更新示意图;
图3是基于排挤策略的多模态蛋白质结构预测方法对蛋白质1hz6预测得到的三维结构。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1~图3,一种基于排挤策略的多模态蛋白质结构预测方法,包括以下步骤:
1)给定输入序列信息,以及蛋白质力场模型,即能量函数rosettascore3;
2)初始化:迭代rosetta协议第一、二阶段,产生具有np个构象的种群pg,记为
3)通过差分进化算法的交叉、变异操作生成种群pg的试验构象种群ug,记为
3.1)从种群pg中选定目标个体
3.2)在[0,l-9]内生成均匀随机整数rand1、rand2和rand3,其中l表示氨基酸序列长度;
3.3)将
3.4)i=i+1,循环步骤3.1)-3.4)直至生成当前种群的试验个体种群ug;
4)为种群pg中的每一个构象
4.1)对ug中的每一个试验构象
4.2)t=t+1,重复步骤4.1),直至t=np,记此时
4.3)将
4.4)i=i+1,循环步骤4.1)-4.4)直至为每一个构象
5)对每一个
其中
6)排挤操作:用
7)聚类操作:生成当前第g+1代种群的模态构象集合
7.4)mg+1初始化为只有一个构象的集合,该构象为种群pg+1中的最好构象;
7.5)将
其中
7.6)i=i+1,循环步骤7.1)-7.3)直至为第g+1代种群找到所有的模态构象;
8)判断是否满足终止条件,若满足则输出结果并退出,否则g=g+1清空mg+1并且返回步骤3)。
进一步,所述步骤2)中,设置最大迭代次数gmax,所述步骤8)中,对种群执行完步骤3)-7)以后,终止条件为迭代次数g达到预设最大迭代次数gmax,若满足输出结果为
本实施例序列长度为67的α/β折叠蛋白质1hz6为实施例,一种基于排挤策略的多模态蛋白质结构预测方法,其中包含以下步骤:
1)给定输入序列信息,以及蛋白质力场模型,即能量函数rosettascore3;
2)初始化:迭代rosetta协议第一、二阶段,产生具有np个构象的种群pg,记为
3)通过差分进化算法的交叉、变异操作生成种群pg的试验构象种群ug,记为
3.1)从种群pg中选定目标个体
3.2)在[0,l-9]内生成均匀随机整数rand1、rand2和rand3,其中l表示氨基酸序列长度;
3.3)将
3.4)i=i+1,循环步骤3.1)-3.4)直至生成当前种群的试验个体种群ug;
4)为种群pg中的每一个构象
4.1)对ug中的每一个试验构象
4.2)t=t+1,重复步骤4.1),直至t=np,记此时
4.3)将
4.4)i=i+1,循环步骤4.1)-4.4)直至为每一个构象
5)对每一个
其中
6)排挤操作:用
7)聚类操作:生成当前第g+1代种群的模态构象集合
7.1)mg+1初始化为只有一个构象的集合,该构象为种群pg+1中的最好构象;
7.2)将
其中
7.3)i=i+1,循环步骤7.1)-7.3)直至为第g+1代种群找到所有的模态构象;
8)判断是否满足终止条件,若满足则输出结果并退出,否则g=g+1清空mg+1并且返回步骤3)。
进一步,所述步骤2)中,设置最大迭代次数gmax=1000,所述步骤8)中,对种群执行完步骤3)-7)以后,终止条件为迭代次数g达到预设最大迭代次数gmax,若满足输出结果为
以序列长度为67的α/β折叠蛋白质1hz6为实施例,运用以上方法得到了该蛋白质的近天然态构象,最小均方根偏差为
以上阐述的是本发明给出的一个实施例表现出来的优良效果,显然本发明不仅适合上述实施例,在不偏离本发明基本精神及不超出本发明实质内容所涉及内容的前提下可对其做种种变化加以实施。
- 还没有人留言评论。精彩留言会获得点赞!