一种基于两阶段采样的蛋白质结构预测方法与流程
本发明涉及一种生物学信息学、智能优化、计算机应用领域,尤其涉及的是一种基于两阶段采样的蛋白质结构预测方法。
背景技术:
蛋白质是所有生物体的基础,是所有生命活动的重要承担者。蛋白质只有折叠成稳定的三维结构时才可以发挥其生物功能、维持生命活动。因此,预测蛋白质的空间三级结构成为生物领域一项重要课题。
目前实验测定的方法主要有x射线衍射、核磁共振和冷冻电镜等三种方法,但这些方法耗时长,费用昂贵,因此在氨基酸序列在指数增长的情况,实验测定的结构只占其中的0.16%。在理论探索和应用需求的双重推动下,根据anfinsen法则,利用计算机设计适当的算法,以序列为起点,三维结构为目标的蛋白质结构预测自20世纪末蓬勃发展。根据预测的难易程度,目标蛋白划分为基于模板和无模板两类。基于模板类目标蛋白在pdb数据库中能够通过序列比对或穿线法识别出一个与目标蛋白序列或结构相似的模板,建模方法主要包括同源建模法和穿线法;无模板类目标蛋白无法通过序列比对或穿线法在现有pdb库中找到满意的结构模板,只能通过从头预测方法建立蛋白结构模型,即直接基于anfinsen法则建立蛋白质物理或知识能量模型,然后,再通过构象搜索优化方法得到目标蛋白的天然结构。
蛋白质结构从头预测空间优化方法一直受到计算生物学领域和进化计算社区的高度关注,是一个热点研究课题。目前常用的蛋白质构象空间优化方法有分子动力学模拟、蒙特卡罗算法以及进化算法等方法。上述所述的这些方法虽然在解决一些小蛋白方面取得了显著的成果,随着序列的增长构象空间的复杂度与多样性也成指数上升,这些方法的搜索能力与采样效率受到很大的限制。最新研究表明,通过两阶段的变异采样,可以有效的提高全局探测和局部增强能力,进而能够得到数量更多、能量更低的构象解,如何设计全局搜索策略与局部扰动策略以及将其有机地结合从而提高求解效率和可靠性将是解决问题的关键。
因此,现有的构象空间搜索方法在预测精度和采样效率方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有的蛋白质结构预测方法存在全局探测与局部增强不平衡,预测精度较低的不足,本发明提出一种采样效率较高、预测精度较高的基于两阶段采样的蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于两阶段采样的蛋白质结构预测方法,所述方法包括以下步骤:
1)给定输入序列信息,选择rosettascore3为能量函数e(x);
2)参数初始化:设置种群规模np,初始构象迭代次数iter,最大迭代次数gmax,变异连续拒绝最大值cmax,状态估计阈值h,温度常数kt,温度变化值t;
3)初始化种群:启动np条montecarlo轨迹,每条轨迹搜索iter次,即生成np个初始个体;
4)对种群中每个个体
4.1)对个体
4.1.1)利用能量函数计算目标个体
4.1.2)对个体
4.1.3)根据montecarlo机制判断是否接收个体
4.1.4)根据公式p=e-δe/kt计算接受概率p,如果p>random,则接受个体
4.1.5)如果连续拒绝了cmax次,则增大温度常数,即kt=kt+t,否则温度常数保持不变;
4.2)对目标个体
4.3)如果i=np,则进入步骤5)进入状态估计,否则转至步骤4.1);
5)根据相邻两代种群的信息,进行状态估计,过程如下:
5.1)计算相邻两代对应个体
5.2)根据公式
6)设置迭代计数器g=0;
7)对每个个体
7.1)利用能量函数计算目标个体
7.2)随机选择一个loop区域,并且在该区域进行3片段组装,生成新个体
7.3)计算目标个体
7.4)如果连续拒绝cmax次,如果是则增大温度常数,即kt=kt+t,否则温度常数保持不变;
7.5)对目标个体
8)判断是否满足终止条件g>gmax,若满足终止条件,则停止迭代,否则进入下一代,返回步骤7)。
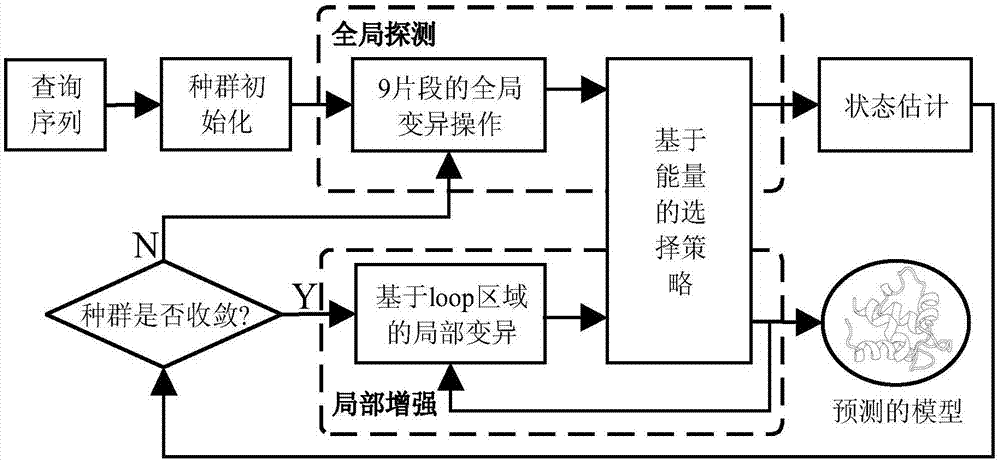
本发明的技术构思为:首先,对种群中的每个个体进行全局的9片段组装,使其形成稳定的三维结构;然后,根据相邻两代间的种群信息进行状态估计,完成全局探测与局部增强的动态切换;最后,对每个个体的loop区域进行局部增强,使其形成更加紧密的三维结构。
本发明的有益效果表现在:一方面,通过两阶段的变异操作使其快速有效的形成紧密的三维结构,进而加快了收敛速度与采样效率;另一方面,利用状态估计模型指导全局探测阶段与局部增强阶段的动态切换,进而大大提高了预测精度。
附图说明
图1是基于两阶段采样的蛋白质结构预测方法的基本示意图。
图2是基于两阶段采样的蛋白质结构预测方法对蛋白质4icb进行结构预测时的构象更新示意图。
图3是基于两阶段采样的蛋白质结构预测方法对蛋白质4icb进行结构预测得到的三维结构图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1~图3,一种基于两阶段采样的蛋白质结构预测方法,所述方法包括以下步骤:
1)给定输入序列信息,选择rosettascore3为能量函数e(x);
2)参数初始化:设置种群规模np,初始构象迭代次数iter,最大迭代次数gmax,变异连续拒绝最大值cmax,状态估计阈值h,温度常数kt,温度变化值t;
3)初始化种群:启动np条montecarlo轨迹,每条轨迹搜索iter次,即生成np个初始个体;
4)对种群中每个个体
4.1)对个体
4.1.1)利用能量函数计算目标个体
4.1.2)对个体
4.1.3)根据montecarlo机制判断是否接收个体
4.1.4)根据公式p=e-δe/kt计算接受概率p,如果p>random,则接受个体
4.1.5)如果连续拒绝了cmax次,则增大温度常数,即kt=kt+t,否则温度常数保持不变;
4.2)对目标个体
4.3)如果i=np,则进入步骤5)进入状态估计,否则转至步骤4.1);
5)根据相邻两代种群的信息,进行状态估计,过程如下:
5.1)计算相邻两代对应个体
5.2)根据公式
6)设置迭代计数器g=0;
7)对每个个体
7.1)利用能量函数计算目标个体
7.2)随机选择一个loop区域,并且在该区域进行3片段组装,生成新个体
7.3)计算目标个体
7.4)如果连续拒绝cmax次,如果是则增大温度常数,即kt=kt+t,否则温度常数保持不变;
7.5)对目标个体
8)判断是否满足终止条件g>gmax,若满足终止条件,则停止迭代,否则进入下一代,返回步骤7)。
本实施例序列长度为76的α折叠蛋白质4icb为实施例,一种两阶段的蛋白质结构预测方法,其中包含以下步骤:
1)给定输入序列信息,选择rosettascore3为能量函数e(x);
2)参数初始化:设置构象集规模np=200,初始构象迭代次数iter=2000,最大迭代次数gmax=100,变异连续拒绝最大计数值cmax=150,状态估计值
3)初始化种群:启动np条montecarlo轨迹,每条轨迹搜索iter次,即生成np个初始个体;
4)对种群中每个个体
4.1)对个体
4.1.1)利用能量函数计算目标个体
4.1.2)对个体
4.1.3)根据montecarlo机制判断是否接收个体
4.1.4)根据公式p=e-δe/kt计算接受概率p,如果p>random,则接受个体
4.1.5)如果连续拒绝了cmax次,则增大温度常数,即kt=kt+t,否则温度常数保持不变;
4.2)对目标个体
4.3)如果i=np,则进入步骤5)进入状态估计,否则转至步骤4.1);
5)根据相邻两代种群的信息,进行状态估计,过程如下:
5.1)计算相邻两代对应个体
5.2)根据公式
6)设置迭代计数器g=0;
7)对每个个体
7.1)利用能量函数计算目标个体
7.2)随机选择一个loop区域,并且在该区域进行3片段组装,生成新个体
7.3)计算目标个体
7.4)如果连续拒绝cmax次,如果是则增大温度常数,即kt=kt+t,否则温度常数保持不变;
7.5)对目标个体
8)判断是否满足终止条件g>gmax,若满足终止条件,则停止迭代,否则进入下一代,返回步骤7)。
以序列长度为76的α折叠蛋白质4icb为实施例,运用以上方法得到了该蛋白质的近天然态构象,最小均方根偏差为
以上说明是本发明以4icb蛋白质为实例所得出的预测效果,并非限定本发明的实施范围,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!