一种基于残基接触信息交叉策略的蛋白质结构预测方法与流程
本发明涉及一种生物学信息学、智能优化、计算机应用领域,尤其涉及的是一种基于残基接触信息交叉策略的蛋白质结构预测方法。
背景技术:
蛋白质是所有生物中最重要的大分子之一,一个细胞的干重的一半以上是蛋白质。蛋白质分子是由氨基酸首尾相连缩合而成的共价多肽链,但是天然蛋白质分子并不是走向随机的松散多肽链。每一种天然蛋白质都有自己特有的空间结构或称三维结构,这种三维结构通常被称为蛋白质的构象,即蛋白质的结构。蛋白质结构调节生物体中的细胞活动,以保持生命。蛋白质分子在分子水平上具体体现了蛋白质结构和功能之间的显著关系。蛋白质在生物学中有许多不同的功能。蛋白质的三维天然结构决定它的生物学机制,而反过来,蛋白质氨基酸单体的一维链又对它的天然结构进行编码。蛋白质的三维结构在药物设计、蛋白质工程和生物技术中具有决定性的重要,因此,蛋白质结构预测是一个重要的研究问题。
由于高通量测序技术的快速发展,加之实验解析蛋白质三维结构技术(x射线晶体学、核磁共振光谱、电子显微镜以及冷冻电镜)耗时且代价昂贵,导致已知序列数目和解析出的蛋白质结构数目间的鸿沟呈指数增加。此外,生物学中心法则中的第二遗传密码和蛋白质折叠机理需要研究学者们进行探索。因此,在无全长模板的情况下(序列相似度<20%),利用计算机,结合优化算法,从氨基酸序列开始,从头预测蛋白质结构是一个研究方法。anfinsen热力学假说表明:蛋白质的空间结构由氨基酸序列唯一确定;蛋白质的空间结构是稳定的;蛋白质的天然构象处于自由能最低点。根据anfinsen原则,以计算机为工具,运用适当的算法,从氨基酸序列出发直接预测蛋白质的三维结构。
科学家提出了许多从头蛋白质结构预测方法,其预测精度在casp比赛的推动下有了巨大的提高。rosetta,quark在历届casp赛事中表现突出。两者都采用片段组装技术,并构建了基于知识的力场模型。然而,利用随机的片段组装,盲目进行构象空间搜索,容易导致不恰当的搜索方向,浪费计算代价,影响最终的聚类结果。
因此,现有的基于随机片段组装的蛋白质结构预测方法高效构象空间搜索和预测精度方面存在着缺陷,需要改进。
技术实现要素:
为了克服现有的基于随机片段组装的蛋白质结构预测方法高效构象空间搜索和预测精度方面的不足,本发明提供一种可以有偏构象空间搜索的基于残基接触信息交叉策略的蛋白质结构预测方法。
本发明解决其技术问题所采用的技术方案是:
一种基于残基接触信息交叉策略的蛋白质结构预测方法,所述方法包括以下步骤:
1)给定输入序列信息,利用robetta服务器获得该序列的片段库;
2)利用raptorx-contact预测该序列的接触信息,并且记录接触概率大于0.6的残基对,假设有n个残基对的接触概率满足该条件,并记第k个残基对间的接触,接触指cα-cα欧氏距离
3)初始化:种群规模np,根据输入序列,最大迭代次数分别为g,执行rosettaabinitio协议的第一与第二阶段np次,产生初始构象种群p={c1,c2,...,cnp},其中cnp表示第np个个体,记当前代数g=0;
4)遍历初始种群的所有个体cm,m∈{1,..np},作为目标个体ctarget进入种群进化的片段组装阶段,并计算当前目标个体的n个残基对的接触情况,tk=1表示第k对残基对接触,tk=0表示第k对残基对不接触;
5)片段组装阶段,过程如下:
5.1)对当前目标个体不接触的残基对,找出接触图中接触概率最大的不接触残基对r,r∈{1,...,n};
5.2)从当前种群中选择一个不同于当前目标个体的个体crandom,该个体需要满足dr,random<dr,target,dr,random和dr,target分别表示crandom和ctarget的第r个残基对的cα-cα欧氏距离;如果当前种群所有个体不满足该条件,则执行下一步;否则执行步骤5.4);
5.3)对目标个体进行随机的片段组装,随机选择窗口,并从对应的片段库中随机选择片段得到测试个体ctrial
5.4)crandom与ctarget交叉:与若jr-ir>l/2,则从crandom的第ir号残基到jr号残基中随机选取长l/2的连续区域,作为片段,其中l是目标序列长度;否则,直接选择crandom的第ir号残基到jr号残基区域作为片段,并将该片段替换到ctarget对应位置得到测试个体ctrial;
5.5)若jr-ir≥9对测试个体ctrial的被组装区域进行长度为9的片段组装,否则进行长度为3的片段组装;随机从被组装区域选择窗口,并从对应的片段库中随机选择片段,得到新的个体c′trial;
5.6)利用能量函数rosettascore3评价c′trial和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受c′trial,若接受,则令ctrial=c′rial;
5.7)重复步骤5.4)-5.5)150次,若metropolis准则拒绝接受150次,则目标个体不被测试个体替换;否则,替换目标个体,令ctarget=ctrial;
6)若g=0,对所有个体执行步骤5),得到下一代种群,并令g=g+1;否则执行步骤8);
7)loop区域微调阶段,过程如下:
7.1)随机选择目标个体loop区域的片段组装窗口,片段长度为3;
7.2)计算片段间的二面角差值平方根,比对对应窗口中片段库中每个片段与当前目标个体该区域的结构相似度,选出片段库中与目标个体当前区域最相似的片段;
7.3)使用选择的片段对目标个体进行片段组装,得到测试个体ctrial;
7.4)利用能量函数rosettascore3评价ctarget和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受ctrial,若接受,则令ctarget=ctrial;
8)遍历当前种群所有个体cm,计算cm的n个残基对的接触情况,若n个残基对的接触,则执行步骤6),否则执行5);得到下一代种群,令g=g+1,判断是否满足终止条件g>g,若满足,结束种群进化,进入下一步;否则重复当前步骤;
9)利用聚类工具spicker对metropolis准则接受的所有过程点聚类,以最大类的类心构象为最终预测结果。
本发明的技术构思为:首先,利用robetta和raptorx-contact得到片段库和接触图;其次,利用接触图信息使得种群个体动态进入片段组装阶段和loop区域微调阶段,并使用了特定测试个体和特定区域的交叉策略;最后,通过聚类得到最终预测结果。
本发明的有益效果表现在:一方面,利用接触图信息,动态进行种群进化;另一方面,利用特定区域的交叉策略,提高搜索效率,提高预测精度。
附图说明
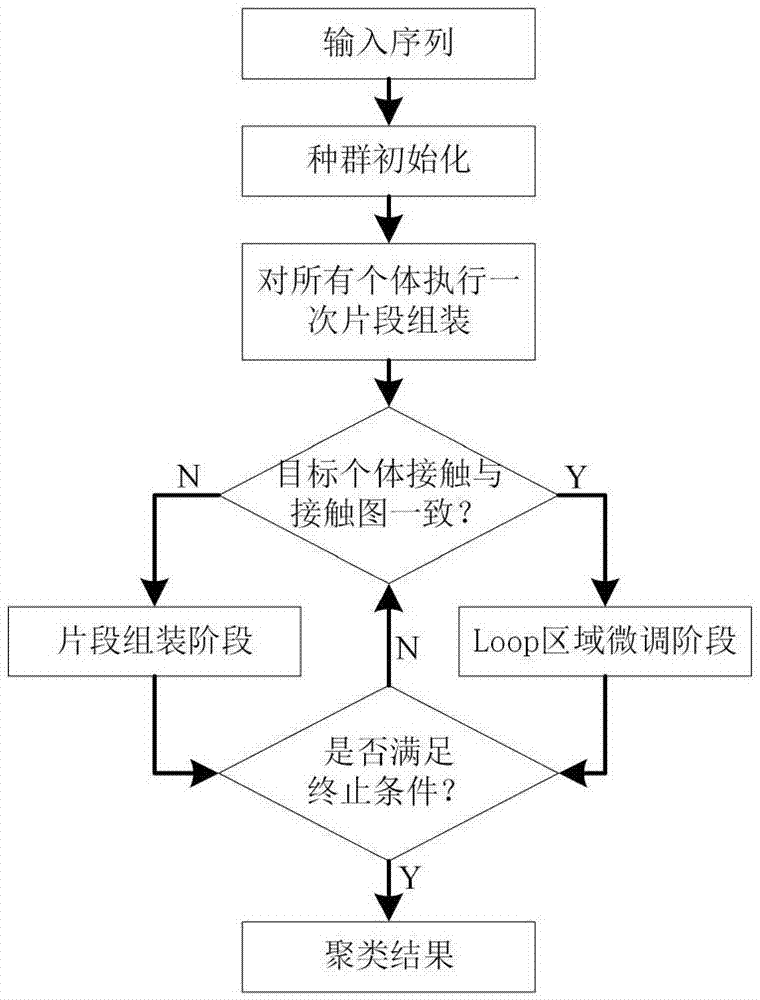
图1是基于残基接触信息交叉策略的蛋白质结构预测方法的基本流程图。
图2是基于残基接触信息交叉策略的蛋白质结构预测方法中对蛋白质1di2a进行结构预测过程中metropolis标准接受构象的能量与天然态对比的rmsd分布。
图3是基于残基接触信息交叉策略的蛋白质结构预测方法对蛋白质1di2a进行结构预测得到的三维结构图。
具体实施方式
下面结合附图对本发明作进一步描述。
参照图1~图3,一种基于残基接触信息交叉策略的蛋白质结构预测方法,包括以下步骤:
1)给定输入序列信息,利用robetta服务器获得该序列的片段库;
2)利用raptorx-contact预测该序列的接触信息,并且记录接触概率大于0.6的残基对,假设有n个残基对的接触概率满足该条件,并记第k个残基对间的接触,接触指cα-cα欧氏距离
3)初始化:种群规模np,根据输入序列,最大迭代次数分别为g,执行rosettaabinitio协议的第一与第二阶段np次,产生初始构象种群p={c1,c2,...,cnp},其中cnp表示第np个个体,记当前代数g=0;
4)遍历初始种群的所有个体cm,m∈{1,..np},作为目标个体ctarget进入种群进化的片段组装阶段,并计算当前目标个体的n个残基对的接触情况,tk=1表示第k对残基对接触,tk=0表示第k对残基对不接触;
5)片段组装阶段,过程如下:
5.1)对当前目标个体不接触的残基对,找出接触图中接触概率最大的不接触残基对r,r∈{1,...,n};
5.2)从当前种群中选择一个不同于当前目标个体的个体crandom,该个体需要满足dr,random<dr,target,dr,random和dr,target分别表示crandom和ctarget的第r个残基对的cα-cα欧氏距离;如果当前种群所有个体不满足该条件,则执行下一步;否则执行步骤5.4);
5.3)对目标个体进行随机的片段组装,随机选择窗口,并从对应的片段库中随机选择片段得到测试个体ctrial
5.4)crandom与ctarget交叉:与若jr-ir>l/2,则从crandom的第ir号残基到jr号残基中随机选取长l/2的连续区域,作为片段,其中l是目标序列长度;否则,直接选择crandom的第ir号残基到jr号残基区域作为片段,并将该片段替换到ctarget对应位置得到测试个体ctrial;
5.5)若jr-ir≥9对测试个体ctrial的被组装区域进行长度为9的片段组装,否则进行长度为3的片段组装;随机从被组装区域选择窗口,并从对应的片段库中随机选择片段,得到新的个体c′trial;
5.6)利用能量函数rosettascore3评价c′trial和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受c′trial,若接受,则令ctrial=c′trial;
5.7)重复步骤5.4)-5.5)150次,若metropolis准则拒绝接受150次,则目标个体不被测试个体替换;否则,替换目标个体,令ctarget=ctrial;
6)若g=0,对所有个体执行步骤5),得到下一代种群,并令g=g+1;否则执行步骤8);
7)loop区域微调阶段,过程如下:
7.1)随机选择目标个体loop区域的片段组装窗口,片段长度为3;
7.2)计算片段间的二面角差值平方根,比对对应窗口中片段库中每个片段与当前目标个体该区域的结构相似度,选出片段库中与目标个体当前区域最相似的片段;
7.3)使用选择的片段对目标个体进行片段组装,得到测试个体ctrial;
7.4)利用能量函数rosettascore3评价ctarget和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受ctrial,若接受,则令ctarget=ctrial;
8)遍历当前种群所有个体cm,计算cm的n个残基对的接触情况,若n个残基对的接触,则执行步骤6),否则执行5);得到下一代种群,令g=g+1,判断是否满足终止条件g>g,若满足,结束种群进化,进入下一步;否则重复当前步骤;
9)利用聚类工具spicker对metropolis准则接受的所有过程点聚类,以最大类的类心构象为最终预测结果。
本实施例序列长度为69的α/β折叠蛋白质1di2a为实施例,一种基于残基接触信息交叉策略的蛋白质结构预测方法,包括以下步骤:
1)给定输入序列信息,利用robetta服务器获得该序列的片段库;
2)利用raptorx-contact预测该序列的接触信息,并且记录接触概率大于0.6的残基对,有n=114个残基对的接触概率满足该条件,并记第k个残基对间的接触,接触指cα-cα欧氏距离
k∈{1,...,n},并记第k个残基对的残基索引号ik和jk,并满足ik<jk;
3)初始化:种群规模np=300,根据输入序列,最大迭代次数分别为g=3000,执行rosettaabinitio协议的第一与第二阶段np次,产生初始构象种群p={c1,c2,...,cnp},其中cnp表示第np个个体,记当前代数g=0;
4)遍历初始种群的所有个体cm,m∈{1,..np},作为目标个体ctarget进入种群进化的片段组装阶段,并计算当前目标个体的n个残基对的接触情况,tk=1表示第k对残基对接触,tk=0表示第k对残基对不接触;
5)片段组装阶段,过程如下:
5.1)对当前目标个体不接触的残基对,找出接触图中接触概率最大的不接触残基对r,r∈{1,...,n};
5.2)从当前种群中选择一个不同于当前目标个体的个体crandom,该个体需要满足dr,random<dr,target,dr,random和dr,target分别表示crandom和ctarget的第r个残基对的cα-cα欧氏距离;如果当前种群所有个体不满足该条件,则执行下一步;否则执行步骤5.4);
5.3)对目标个体进行随机的片段组装,随机选择窗口,并从对应的片段库中随机选择片段得到测试个体ctrial
5.4)crandom与ctarget交叉:与若jr-ir>l/2,则从crandom的第ir号残基到jr号残基中随机选取长l/2的连续区域,作为片段,其中l=69是目标序列长度;否则,直接选择crandom的第ir号残基到jr号残基区域作为片段,并将该片段组装到ctarget对应位置得到测试个体ctrial;
5.5)若jr-ir≥9对测试个体ctrial的被组装区域进行长度为9的片段组装,否则进行长度为3的片段组装;随机从被组装区域选择窗口,并从对应的片段库中随机选择片段,得到新的个体c′trial;
5.6)利用能量函数rosettascore3评价c′trial和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受c′trial,若接受,则令ctrial=c′trial;
5.7)重复步骤5.4)-5.5)150次,若metropolis准则拒绝接受150次,则目标个体不被测试个体替换;否则,替换目标个体,令ctarget=ctrial;
6)若g=0,对所有个体执行步骤5),得到下一代种群,并令g=g+1;否则执行步骤8);
7)loop区域微调阶段,过程如下:
7.1)随机选择目标个体loop区域的片段组装窗口,片段长度为3;
7.2)计算片段间的二面角差值平方根,比对对应窗口中片段库中每个片段与当前目标个体该区域的结构相似度,选出片段库中与目标个体当前区域最相似的片段;
7.3)使用选择的片段对目标个体进行片段组装,得到测试个体ctrial;
7.4)利用能量函数rosettascore3评价ctarget和ctrial,得到能量值e′和e,并根据metropolis准则选择是否接受ctrial,若接受,则令ctarget=ctrial;
8)遍历当前种群所有个体cm,计算cm的n个残基对的接触情况,若n个残基对的接触,则执行步骤6),否则执行5);得到下一代种群,令g=g+1,判断是否满足终止条件g>g,若满足,结束种群进化,进入下一步;否则重复当前步骤;
9)利用聚类工具spicker对metropolis准则接受的所有过程点聚类,以最大类的类心构象为最终预测结果。
以序列长度为69的α/β折叠蛋白质1di2a为实施例,运用以上方法得到了该蛋白质的近天然态构象,最小均方根偏差为
以上说明是本发明以1di2a蛋白质为实例所得出的预测效果,并非限定本发明的实施范围,在不偏离本发明基本内容所涉及范围的前提下对其做各种变形和改进,不应排除在本发明的保护范围之外。
- 还没有人留言评论。精彩留言会获得点赞!