一种煤微观孔隙结构表征方法与流程
本发明属于煤微观孔隙结构测量技术领域,涉及一种煤大分子间孔隙的表征及孔隙度的测量方法,尤其涉及一种利用煤大分子与分子模拟技术对煤微观模型孔隙结构的表征测试方法。
背景技术:
煤是典型的多孔介质之一,易于吸附瓦斯等小分子物质。在煤的瓦斯吸附过程中,煤中孔隙的存在对瓦斯的吸附起到重要作用。国内外学者对煤的孔隙结构、孔隙率等进行研究,多种手段被应用于研究煤的孔隙,以比重法、等温吸附法、压汞法为主。但由于煤内部结构复杂及测量尺度的限制,这些测量煤孔隙的方法难以较为准确地反映煤内部微观孔隙结构。由于煤大分子具有高度立体化的特点,它的结构特点决定了煤中存在大量的细微孔隙,而微孔隙中具有的可供小分子吸附的表面积也是十分巨大的,因此煤中大分子间形成的微孔隙对于瓦斯吸附具有重要的作用。本发明主要从表征煤大分子间孔隙出发,提出了一种新的描述煤微观孔隙结构的方法,丰富了目前煤微观孔隙结构的表征参数。
技术实现要素:
为了克服现有煤微观孔隙表征方法的不足,本发明提供一种基于分子模拟技术分析煤大分子之间微孔隙的表征方法,应用实验及分子模拟相结合的方法构建煤大分子结构。通过蒙特卡洛方法构建煤微观模型,并对其进行优化。利用“滚球方法”对煤微观模型内部的孔隙结构进行定量分析,本发明适用于煤内部大分子间孔隙的测定与表征。
本发明解决其技术问题所采用的技术方案是:
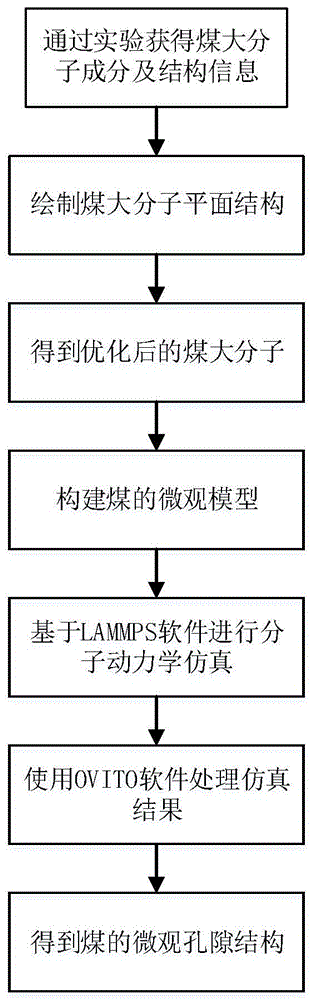
一种煤微观孔隙结构表征方法,其特征在于:本方法基于分子模拟软件materialsstudio和lammps建立煤的微观模型,并对其进行分子间孔隙表征,包括如下步骤:
步骤1、进行相关实验得到煤大分子的成分组成及结构信息;
步骤2、利用步骤1中得到的实验数据在chemdraw和mestrevona软件中绘制煤大分子模型,导出到materialsstudio软件中并对其进行优化;
步骤3、基于步骤2中得到的煤大分子模型,应用蒙特卡洛方法构建由多个煤大分子组成的微观模型,并将该模型通过ubuntu导入到lammps软件中;
步骤4、通过lammps的fix命令对煤的微观模型进行相应的参数设定,进行能量最小化、退火及分子动力学优化;
步骤5、根据需要在lammps中通过dump命令得到基体内原子坐标文件;
步骤6、基于ovito软件对步骤5中得到的煤微观模型坐标文件进行数据处理,并对煤微观模型中原子的运动轨迹进行可视化,得到目标煤微观模型的孔隙信息;
本发明所述的煤微观孔隙结构表征方法与传统煤孔隙测量方法相比,从分子尺度考虑了煤中存在的微孔隙,考虑煤大分子结构对其内部孔隙的本质影响,对于表征真实情况下煤微孔隙具有实际意义。
作为本发明的一种技术方案,所述步骤1中煤大分子结构的构建实验方法包括:实验样品制备、通过元素分析和工业分析实验、核磁共振实验、x射线光电子能谱实验、傅里叶变换红外光谱实验、x射线衍射实验得到构建煤分子的成分组成及分子结构信息。
作为本发明的一种技术方案,所述步骤2中对煤大分子进行优化的方法包括:
在materialsstudio软件中的pcff分子力场下选择forcite模块中的smartminimizer算法对煤大分子结构进行结构优化。参数设置:迭代步数50000步:截断半径15.5a,能量停止容差为0.001kcal/mol,力的停止容差为0.5kcal/mol/a。采用电荷平衡法,基于原子状态对库伦能和范德华能进行计算。
为了克服分子结构能垒,使体系的能量达到几何最优状态,采用退火动力学计算,初始温度设置为300k,最高温度设置为500k,升温速率为600k/ps,选择nvt系综,模拟时间是10ps,控温方法选择nose方法,退火模拟的循环次数设置为5,在每次循环结束后对输出的构型进行几何优化,使体系的能量和结构达到最优状态。
作为本发明的一种技术方案,所述步骤3中应用蒙特卡洛方法构建煤基体模型包括:
利用构建及优化完成的煤大分子模型,选择materialsstudio软件中的amophouscellconstruction模块对煤大分子添加周期性边界条件,通过改变煤大分子晶胞的体积,获得煤大分子晶胞势能随密度变化的变化规律。
煤大分子第一次达到能量最低点时得到的模拟密度与真实密度存在较大差距,超越第一次能量最低值后的能量变化的最低值对应的晶胞密度与煤的真实密度才最为接近根据计算出的最优晶胞密度,以此为依据构建含有多个煤大分子的分子聚集体模型。
作为本发明的一种技术方案,所述步骤为4中对煤微观模型进行退火及分子动力学优化包括:
利用步骤3中将构建好的含多个煤大分子的聚集体模型导出为lammps可以运行的data文件。在npt系综下进行动力学优化,系统保持压力0.1mpa,温度设定为300k,分别选择berendsen和nose方法对系统压力和温度进行控制。时间步长为1fs,截断半径设置为15.5a,优化时间300ps,得到符合最优情况下的煤微观模型的密度值。
为了避免煤微观模型中分子堆积等不合理构象产生的局部能量过大的情况,需要对其进行构型优化及退火动力学处理。选择共轭梯度法对基体进行能量最小化,能量停止容差为0.001kcal/mol,力的停止容差为0.5kcal/mol/a,最大迭代步数为50000步,采用ewald算法对长程库伦相互作用进行计算。退火动力学处理与上面提到的方法一致,退火完毕后在nvt系综下积分100ps得到驰豫后的煤微观模型。
作为本发明的一种技术方案,所述步骤为6中求取目标煤基体的孔隙信息及输出表征构象包括;
通过步骤5输出煤大分子微观模型最终构型的坐标文件,运用ovito软件中的delaunay四面体剖分算法将煤微观模型空间划分为四面体单元,利用基于alpha-shape算法的“滚球方法”对煤微观模型内部孔隙进行探测,得到相应的内部孔隙结构。
附图说明
图1为本发明的煤微观孔隙结构表征方法的流程图。
图2为本发明通过实验数据绘制出的煤大分子平面结构
图3为本发明经过结构优化及退火处理后的煤大分子最优构型
图4为本发明煤大分子晶胞不同密度对应势能的分析图
图5为本发明不同直径探针下的煤微观模型孔隙结构图
具体实施方式
为使本发明的上述和其他目的、特征和优点能更明显易懂,下文特举出赵庄煤微观孔隙结构表征的实施例,并配合图示,做详细说明如下。
如图1所示,图1为本发明的煤微观孔隙结构表征方法的流程图。
在步骤1中,构建赵庄煤大分子模型需要的样品制备方法及实验如下:
依据《煤层煤样采取方法》(gb/t482-2008)进行采样,并将新采集的样品按一定要求包装。将样品粉碎、研磨,然后在60-80目的范围内筛分,用氢氟酸和盐酸等无机酸进行脱矿处理,以减少某些无机矿物质对实验测试结果的影响。用蒸馏水清洗至中性,后在真空干燥箱100℃下干燥12小时直至恒重。
元素分析和工业分析实验对水分、灰分、和挥发分三个参数进行工业分析,利用5e-macⅲ快速分析仪采集数据。元素分析,包括对c、h、n和s的分析,在flash2000元素分析仪上进行,该分析仪的分析原理基于经典的pregl和dumas方法。利用差减法求出o的含量。
核磁共振实验利用布鲁克avanceⅲ型的超导核磁共振仪器进行核磁共振测试实验。测试频率600mhz,脉冲12.95μs,扫描次数128次。实验采用固体双共振探头,采样时间0.05s,zro2型号转子的外径6mm,角度转速为6-7khz。
x射线光电子能谱实验在coretech公司生产的能谱仪上进行xps测试。相关参数为单色aika(hv=1486.6ev),功率150w,650μm束斑;电荷校正采用碳c1s=284.8ev进行校正;通能窄扫20ev,宽扫100ev,真空度1×10-10mbar,输出功率300w。
傅里叶变换红外光谱实验在nicoletnexus410型红外光谱仪上进行测试。波数范围是400-4000cm-1。光谱仪分辨率2cm-1,信噪比是50000:1,扫描32次。
x射线衍射实验在d8advance衍射仪上完成测试,采用铝框架制样法,管电流40ma,管电压40kv,cu靶波长是1.5406a,co靶波长是1.79026a。扫描范围2θ=5°-8°,ds=1°,ss=1°,rs=0.15mm,扫描速度ω=8°/min,强度单位为cps(计数/s)。通过以上实验数据及分峰拟合获得构建煤大分子的成分组成及结构信息,得到相应的赵庄煤大分子模型。
在步骤2中,利用步骤1中得到的实验数据在chemdraw和mestrevona软件中绘制煤大分子模型,如图2所示。将其导出到materialsstudio软件中并对其进行优化。对构建完成的赵庄煤大分子模型选择的能量最小化方法为smartminimizer方法,该方法可以混合最速下降法、共轭梯度法和牛顿法进行结构优化,具有单独使用上述方法所不具备的优势。在pcff分子力场下选择forcite模块中的smartminimizer方法对煤大分子结构进行结构优化。参数设置:迭代步数50000步:截断半径15.5a:能量停止容差为0.001kcal/mol,力的停止容差为0.5kcal/mol/a。采用电荷平衡法:基于原子状态对库伦能和范德华能进行计算。为了克服分子结构能垒,使体系的能量达到最优状态,所以进行退火动力学计算,初始温度设置为300k,最高温度设置为500k,升温速率为600k/ps,选择nvt系综,模拟时间是10ps,控温程序选择nose方法,退火模拟的循环次数设置为5,在每次循环结束后对输出的构型进行几何优化,最终优化构型如图3所示。
在步骤3中,根据步骤2中得到的赵庄煤大分子优化模型,首先对一个赵庄煤大分子模型添加周期性边界条件,通过改变煤分子晶胞的体积,获得含有一个煤大分子晶胞势能随煤大分子晶胞密度的势能变化规律,如图4所示。选择超越第一次能量最低值后的能量变化的最低值对应的晶胞密度与煤的真实密度才最为接近。
得到符合最优情况的赵庄煤大分子晶胞密度为1.15g/cm3,基于蒙特卡洛方法,选取20个优化好的赵庄煤大分子模型,在materialsstudio中的amophouscellconstruction模块中建立含有20个赵庄煤大分子的分子聚集体。
为了提高计算效率及后续孔隙计算,需要将基体模型导出为lammps可以运行的data文件,具体步骤为将构建好的含有20个煤大分子的微观模型在materialsstudio中导出为car文件。在lammps的msi2lmp/frc_files文件夹下执行make命令,将会生成msi2mp.exe可执行文件,把car文件和msi2mp.exe文件放在lammps力场文件下输入命令./msi2lmp.execoal-classii-frcpcff>coal.data,生成的coal.data就是需要的data文件,data文件中包含赵庄煤微观模型的原子坐标、结构信息、及力场参数等相关数据。
在步骤4中,根据步骤3中得到的赵庄煤微观模型,对其进行能量最小化及退火动力学驰豫。
首先将构建好的赵庄煤微观模型在npt系综下进行动力学优化,系统保持压力0.1mpa,温度设定为300k,分别选择berendsen和nose方法对压力和温度进行控制。时间步长为1fs,截断半径设置为15.5a,模拟时间300ps,模型最终尺寸为4.25nm×4.25nm×4.25nm,得到符合真实情况下的模型密度为1.153g/cm3。与步骤3中的最优化煤分子晶胞密度接近。
为了避免基体模型中分子堆积等不合理构象产生的局部能量过大的情况,需要对其进行构型优化及退火动力学处理。选择共轭梯度法对基体进行能量最小化,能量停止容差为0.001kcal/mol,力的停止容差为0.5kcal/mol/a,最大迭代步数为50000步,采用ewald算法对长程库伦相互作用进行计算。退火动力学处理与上文提到的方法一致,退火完毕后将赵庄煤的微观模型在nvt系综下保持温度300k,积分100ps得到驰豫后的稳定结构。
在步骤5中利用lammps中的dump命令将驰豫后的模型导出为lammpstrj文件。
在步骤6中根据步骤5得到的赵庄煤微观模型,应用ovito中的delaunay四面体剖分算法将其空间划分为四面体单元,并利用基于alpha-shape算法的“滚球方法”对基体内部孔隙进行探测,分别选择直径0.4nm,0.5nm,0.6nm的探针对其进行孔隙探测,得到相应的内部孔隙结构,如图5所示。对应的命令如下所示。
fromovito.ioimportimport_file
fromovito.modifiersimportconstructsurfacemodifier
node=import_file("simulation.dump")
mod=constructsurfacemodifier(radius=*)
node.modifiers.append(mod)
node.compute()
print("surfacearea:%f"%node.output.attributes['constructsurfacemesh.surface_area'])
print("solidvolume:%f"%node.output.attributes['constructsurfacemesh.solid_volume'])
fraction=node.output.attributes['constructsurfacemesh.solid_volume']/node.output.cell.volume
print("solidvolumefraction:%f"%fraction)
mesh=node.output.surface
mesh.export_vtk('surface.vtk',node.output.cell)
- 还没有人留言评论。精彩留言会获得点赞!