构建结直肠肿瘤状态评估模型的方法及应用与流程
本发明涉及基因检测及生物信息学技术领域,具体涉及一种基于高通量测序数据和临床表型构建结直肠癌状态评估方法,及其相关检测panel设计和实施应用案例。
背景技术:
第一代测序技术,通过双脱氧末端终止法或化学切割法,获得序列特定位置的碱基信息,利用电泳和显影读取核酸序列。基因芯片技术,通过与一组已知序列的核酸探针杂交进行核酸序列测定的方法,实现了高通量并行化,缺点在于可重复性和灵敏度有待增强,分析范围不够广泛。二代测序技术,又称新一代测序技术(next-generationsequencing,ngs),与第一代测序不同,通过体外片段扩增和边合成边测序,实现了高通量并行化测序,主要缺点是读长短。第三代测序技术,又称单分子测序技术,无需扩增,直接通过检测模板序列的荧光信号或电信号,不受读长限制地直接读取模板序列信息。高通量测序数据(第二代或第三代测序技术生成),在dna层面可以高通量检测突变,包括点突变、插入缺失突变、基因融合、拷贝数变异等,在rna层面则可高通量检测基因定量表达水平、可变基因剪切与融合等,对于精准医学发展起到重要推动作用。
以肿瘤、心脑血管疾病、代谢类疾病为代表的复杂疾病是人类健康的重大威胁,目前对复杂疾病致病机制的研究,得益于生物技术的快速发展,有了长足进步。基于复杂疾病样本的高通量测序数据,可以从分子水平阐释复杂疾病的发生、进展、转归、治疗和预后的规律,辅助有效评估肿瘤状态,为制定精准有效的治疗方案提供指导。肿瘤是复杂疾病的典型代表,其导致的可检测的突变或异常表达基因,同具体肿瘤临床表型密切相关时,即有可能作为分子肿瘤标志物,用于诊断、风险评估、预后、指导治疗、进展和安全性评估等。
基于高通量测序数据的复杂疾病标志物发现技术和相关标志物检测评估方案已经取得长足进步,但仍面临如下不足有待克服:1)标志物挖掘方法相对简单,准确性和可解释性亟需增强。对于多基因相关的复杂疾病,基于单个基因的标志物难以达到高准确性;相对于提高准确性,对标志物的机制可解释性关注更为不足。这既不符合循证医学理念以及理解标志物关键原理,也不利于实现标志物的理论最优组合,从而提高其鲁棒性和可重复性。2)检测和评估内容相对单一,功能有限。目前,由于基因收集筛选能力和测序成本,同一标志物检测方案覆盖的基因相对较少,实际应用中以单位点或小片段突变为主要评估指标,近年来以基因表达水平和检测panel中所有基因整体突变水平作为标志物评估的方案日益受到关注;在功能方面,以位点或基因相关靶向药效果预测为主,对于更广泛的手术、化疗、放疗、免疫治疗等指导意义有限。3)标志物设计及配套数据分析工具,对多元信息的利用不够充分。目前多数设计方案仅针对药物指南、标签以及有限的文献收集,技术路线侧重于单一组学层面,基于大规模测序结果、公共数据库及文本挖掘技术综合分析较少,对涵盖多种分子组学和临床表型信息的多元数据整合分析严重不足。
技术实现要素:
为了解决上述问题,本发明提出了一种基于转录组数据、外显子组/基因组数据和临床表型挖掘结直肠癌标志物的方法,设计了一套整合高通量测序数据和临床表型构建结直肠癌状态评估模型的计算方法,筛选到了结直肠癌相关的生物标志物,形成了相应的疾病状态评估模型。
通过本发明建立的方法挖掘到的结直肠癌标志物,兼顾标志物准确性与机制解释性;本发明所设计的结直肠癌状态评估模型构建方法,对多元信息利用充分,评价指标丰富,功能系统全面实用,涵盖筛选挖掘、建模评分与检测panel设计等。以上技术创新能在结直肠癌标志物挖掘和状态评估模型构建中进行了具体实施。
本发明提出了一种基于转录组数据、外显子组数据和临床表型挖掘复杂疾病标志物的方法,包括以下步骤:
步骤1)对复杂疾病病例信息分类整理:
步骤1.1)将所述复杂疾病病例信息分为转录组数据、外显子组/基因组数据和临床信息;
步骤1.2)将所述复杂疾病病例信息按照疾病状态分类并进行配对整理;以上信息分类将用于步骤2三种方法模式的选择。
步骤2)构建复杂疾病标志物组合,使用基于贪婪算法的逐次迭代和/或基于遗传算法的进化迭代进行组合优化筛选:
若所述复杂疾病病例信息仅涉及转录组数据与临床信息,则执行步骤2.1)基于转录组数据与临床信息进行标志物挖掘,构建复杂疾病相关的基因异常调控关系标志物组合;
若所述复杂疾病病例信息仅涉及外显子组/基因组数据与临床信息,则执行步骤2.2)基于外显子组/基因组数据与临床信息进行标志物挖掘,构建复杂疾病相关的基因变异标志物组合;
若所述复杂疾病病例信息同时包含转录组数据、外显子组/基因组数据与临床信息,则执行步骤2.3)基于转录组数据、外显子组/基因组数据与临床信息进行标志物挖掘,构建复杂疾病相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤2.1)包括以下子步骤:
步骤2.1.1)构建参考基因调控网络:从公共数据资源中可获取的转录调控关系信息以及人类编码基因启动子序列出发,识别潜在的转录因子(transcriptionalfactor,tf)与靶基因(target)关系对,构建参考基因调控网络(referencegeneregulationnetwork,rgrn)。
步骤2.1.2)基于特定疾病状态下的转录组表达数据以及rgrn中的tf-target关系,构建特定疾病状态下的,即条件特异的基因调控网络(conditionalgeneregulationnetwork,cgrn)。步骤2.1.2)中,采用基于机器学习的特征选择算法,包括boruta、
步骤2.1.3)量化条件特异的基因调控网络中的基因调控强度和网络间调控强度差异:采用多元线性回归模型量化条件特异的基因调控网络中的基因调控强度;
采用de-biasedlasso方法进行回归,求解得到每一个基因调控关系的调控强度及其置信区间,通过比较不同条件特异的基因调控网络中同一调控关系的置信区间是否有重叠,判定其调控差异是否显著;或通过比较不同条件特异的基因调控网络中同一调控关系的强度均值变化,无需计算置信区间,直接量化其调控差异。
步骤2.1.4)筛选不同疾病状态下的条件特异的基因调控网络之间的基因异常调控关系:
整合三方面与基因调控相关的因素,筛选不同疾病状态下的条件特异的基因调控网络之间的基因异常调控关系,包括:基因调控强度显著变化、调控目标基因表达水平显著变化,以及tf对target的调控强度变化方向与target表达水平变化方向一致;同时,根据调控强度在不同疾病状态间的差异程度,对筛选到的基因异常调控关系进行排序。
步骤2.1.5)基于基因异常调控关系,构建复杂疾病状态(如疾病进展阶段、预后、治疗方案敏感性)相关的基因异常调控关系标志物组合,该标志物组合可用于疾病进展评估、预后评估、治疗方案辅助决策。
步骤2.1.5从基因异常调控关系出发,采用cox回归模型筛选与疾病状态,如疾病进展阶段、预后、治疗方案敏感性相关的标志物组合。其中,涉及每个异常调控基因对的cox模型构建及其c-index比较、基于贪婪算法的异常调控基因对逐次增加迭代,以及基于遗传算法的进化式迭代。
具体地,所述步骤2.2)包括以下子步骤:
步骤2.2)基于外显子组/基因组数据与临床信息的标志物挖掘;
步骤2.2.1)识别与复杂疾病相关的基因变异;其中,与疾病状态相关的dna变异包括基因拷贝数与体细胞突变,包括但不限于单个碱基多态性(snp)、插入与缺失(indel)、拷贝数变异(cnv)、基因融合(fusion)、基因重排(rearrangement)等高通量测序技术可以检测的变异;
步骤2.2.2)采用数据驱动和/或先验知识驱动定量筛选复杂疾病状态相关的重要基因变异;其中,数据定量过滤筛选,涉及体细胞基因变异频率计算、排序,以及高频变异基因识别,其中基因变异频率≥5%的基因进一步用于先验知识过滤;先验知识过滤筛选,包括应用标准、临床治疗指南、药物标签及通用知识库和文献报道中的复杂疾病相关基因;
步骤2.2.3)基于步骤2.2.2)得到的复杂疾病状态相关的重要基因变异,构建复杂疾病状态(如疾病进展阶段、预后、治疗方案敏感性)相关的dna变异标志物组合,该标志物组合可用于疾病进展评估、预后评估、治疗方案辅助决策。其中,采用cox回归模型筛选与疾病状态,如疾病进展阶段、预后、治疗方案敏感性相关的dna变异标志物组合。其中,涉及每个变异的cox模型构建及其c-index比较、基于贪婪算法的重要变异逐次增加迭代,以及基于遗传算法的进化式迭代。以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建复杂疾病相关的基因变异标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤2.3)包括以下子步骤:
步骤2.3.1)对于同时具备转录组数据和外显子组/基因组数据的复杂疾病数据集,利用步骤2.1.1~2.1.4筛选疾病状态相关的基因异常调控关系,同时利用步骤2.2.1~2.2.2挖掘疾病状态相关的重要基因变异,分别得到复杂疾病相关的基因异常调控关系和重要基因变异;
步骤2.3.2)随后采纳步骤2.1.5和步骤2.2.3中,基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,整合rna和dna信息,构建复杂疾病相关的基因异常调控关系和基因变异标志物组合。
基于上述方法得到的复杂疾病标志物,本发明提出了一种复杂疾病综合状态评分方法,包括以下步骤:
步骤3.1)针对已知先验知识,筛选复杂疾病状态相关的临床信息(如疾病进展阶段、预后、治疗方案敏感性)及检验和病理指标;
步骤3.2)从复杂疾病队列中病例信息出发,筛选复杂疾病状态相关的临床信息及检验和病理指标;
步骤3.3)将本发明方法得到的复杂疾病相关的基因异常调控关系和/或基因变异标志物组合,同步骤3.1和3.2筛选所得复杂疾病状态相关的临床信息及检验和病理指标整合,优化成为复杂疾病多元标志物组合,构建复杂疾病综合状态评分模型;所述模型用于复杂疾病综合状态评分计算。其中,利用基于贪婪算法的逐次增加迭代和/或基于遗传算法的进化迭代,将复杂疾病相关的基因异常调控关系和/或基因变异标志物组合、复杂疾病相关临床信息及检验和病理指标,整合优化精简特征后形成复杂疾病多元标志物组合,包含复杂疾病相关的基因异常调控关系、基因变异、临床信息、检验和病例指标;进而利用统计回归和机器学习算法,针对复杂疾病预后评估、治疗效果预测及治疗方案辅助决策,构建复杂疾病综合状态评分模型。
具体地,步骤3.1采用公开渠道可获得的国内外最新临床指南、专家共识和推荐意见,药物临床应用指南,来自中国临床肿瘤学会(csco)、美国国家综合癌症网络(nccn)、美国临床肿瘤学会(asco)、欧洲肿瘤内科学会(esmo)和日本肿瘤学会(jsc)的临床实践指南,以及通用知识库中的复杂疾病相关各类检验指标,结合复杂疾病相关知名本体库和公开发表的权威文献,系统检索和挖掘同复杂疾病进展、治疗方案敏感性和预后高度关联的检验指标,去除冗余后,纳入后续模型及工具开发。
具体地,步骤3.2基于可以得到的复杂疾病队列数据,整合复杂疾病状态评估相关检验指标和临床信息构建模型,利用预测评估指标(如c-index、auc),采用机器学习特征选择策略,如boruta、
具体地,步骤3.3利用统计建模或机器学习手段,将前文所述测序组学标志物、临床检验指标,以及基于疾病队列信息筛选到的指标,结合病例临床信息,训练实现复杂疾病状态评估模型;并以更为准确可靠地预测复杂疾病患者预后状态和治疗方案受益情况为目标,综合使用各类指标(如生存曲线、c-index、auc等)精简特征组合(最优组合目标是特征数量少,准确可靠,机制可解释性强),迭代优化状态评估模型。
本发明提出了一种复杂疾病综合状态评分计算系统,采用本发明复杂疾病综合状态评分方法,将复杂疾病综合状态评分模型开发封装为方便使用的复杂疾病综合状态评分计算系统(如软件、在线服务器形式)。其中,必须包括实用方便的输入和输出模块和评分模型,其中输出内容应至少包括复杂疾病的分类及风险评分以及对应的治疗受益预测提示等信息。
本发明提出了一种基因检测panel设计方法,包括以下步骤:
步骤4.1)基于本发明方法筛选得到复杂疾病相关的基因异常调控关系和/或基因变异标志物组合,并最终纳入本发明所述复杂疾病综合状态评分方法的基因集,梳理基因集中基因相关信息,去除冗余,确定标准基因名;
步骤4.2)针对步骤4.1)中梳理后的基因,选择用于复杂疾病检测设计的靶基因目标区域,可用于探针设计或引物设计;
步骤4.3)根据步骤4.2)中的靶基因目标区域,设计相应的探针和/或引物序列,并记录重要注释;
步骤4.4)针对步骤4.2)中的靶基因目标区域,参考人类基因组中可设计探针和/或引物数据集,对靶基因目标区域进行优化设计,使探针和/或引物能均匀捕获覆盖目标区域;
步骤4.5)将步骤4.3和4.4中的靶基因目标区域相关探针和/或引物设计区域进行比对,获取具有最优覆盖度的靶基因目标区域相关探针和/或引物设计方案;
步骤4.6)基于步骤4.5设计的靶基因目标区域相关探针和/或引物,制作出用于充分进行复杂疾病状态评估的基因检测panel。
具体地,步骤4.2选择探针设计的基因目标区域时,采取精准优先、逐步扩大的原则,首先采用变异位点区域,次优选择变异位点所在外显子区域,最后可采用变异基因的全部可变剪切区域。其中,选择用于复杂疾病检测的探针和/或引物设计的靶基因目标区域,遵循以下原则:对于变异位点信息明确具体且该变异位点序列前后各100bp范围内无其它变异位点,则将此已明确的基因位点覆盖区域作为靶基因目标区域;对于变异位点较集中或密集的基因区域,即两个变异位点相邻且间隔不超过100bp,则选择该组变异位点的外显子作为靶基因目标区域;对于步骤4.1)确定的信息非常多样的重要基因,在前两项设计不适用的情况下,则选择该基因全部可变剪切类型的区域作为靶基因目标区域。
具体地,步骤4.3)中的设计是指采用对步骤4.2)中的靶基因目标区域两端延伸,合并延伸后的全部目标区域并去除冗余;以合适的文件格式记录探针和/或引物设计的靶基因目标区域的重要信息,包括靶基因目标区域的染色体编号、靶基因目标区域的起始位置、靶基因目标区域的终止位置、突变位点信息、自定义信息,如引物设计所需3’端信息。
具体地,步骤4.4)中参考人类基因组中可设计探针和/或引物数据集,对靶基因目标区域设计的探针和/或引物覆盖深度进行加权,并基于人类全基因组测序数据预测其探针和/或引物覆盖深度后,在全探针和/或引物数据集进行调整,使探针和/或引物能均匀捕获覆盖目标区域。
具体地,步骤4.5综合比对步骤4.3和步骤4.4所生成的探针设计区域,并同时测评探针对重要变异位点和全部目标区域的覆盖度,获取具有最优覆盖度的探针设计方案。其中,步骤4.5)中靶基因目标区域相关探针和/或引物的最优覆盖度是指计算探针和/或引物对步骤4.1)所述重要基因变异位点的覆盖度和全部靶基因目标区域的覆盖度,计算公式为:覆盖度=比对上的读长数/目标测序读长数;通过靶基因目标区域附近的优化,使得最终设计的探针和/或引物,对全部靶基因目标区域的覆盖度≥90%,同时对步骤4.1)所述重要基因变异位点的覆盖度≥97%。
本发明中,步骤4.1至4.6整体上是一个综合流程,可以根据具体检测中采用的检测平台,如pcr、ngs、三代测序、nanostring等。针对不同领域和技术经验规范,可进行相应的调整优化。
本发明提出了一种基于高通量测序数据和临床表型构建复杂疾病状态评估方法,基于复杂疾病状态评估基因检测panel同综合状态评分计算系统的联用进行评估,包括以下步骤:
步骤5.1)基于本发明所述方法设计的基因检测panel,得到复杂疾病相关的基因异常调控关系和/或基因变异标志物组合的定量值,本发明所述的复杂疾病综合状态评分计算系统;
步骤5.2)将获取本发明所述复杂疾病状态相关的临床信息及检验和病理指标的定量值,输入本发明所述的复杂疾病综合状态评分计算系统;
步骤5.3)将步骤5.1)和5.2)所涉及的硬件、软件和/或在线工具,组合为一套配套联用的流程,使得用户根据需求可以完成检测、信息输入、计算评估和结果获取,顺利获得评估状态和提示建议输出等有效信息。
本发明中,步骤5.1采用适应具体应用需求的方式,如检测装置或试剂盒,灵活获取dna和rna层面包括但不限于拷贝数、基因变异和基因表达在内的多种组学信息,以得到输入综合状态评分计算系统的定量数值为目标,并确定规范输入方式。
本发明中,步骤5.2采用适合应用场景,并与步骤5.1中基因检测panel配套的硬件或软件模块,以自动化或人工方式从his或emr等医学信息系统中获取可以输入综合状态评分计算系统的病例检测指标和临床信息,并确定规范输入方式。
本发明中,步骤5.3构建的基因检测panel同综合评分系统的联用方法,以符合应用需求为目标,组合形式灵活多样,包括但不限于试剂盒/软件,检测装置/数据处理一体机,试剂盒/检测装置/数据在线分析平台等形式;使用者可以按说明文档,以最方便友好和高效的形式,输入个体病例的必要信息,包括复杂疾病相关的基因异常调控关系和/或基因变异标志物组合、复杂疾病相关临床信息及检验和病理指标,自动化或半自动化进行数据汇总统计和预处理之后,完成计算评估,并输出该个体病例的分类及风险评分以及对应的治疗受益预测提示等信息;最终可以实现对复杂疾病个体病例状态的评估,辅助临床决策等功能。
本发明所述的方法在构建基于高通量测序数据和临床表型构建复杂疾病状态评估模型中的应用,包括在筛选复杂疾病综合状态评估标志物组合中的应用;在筛选肿瘤综合状态评估标志物组合中的应用;在复杂疾病预后评估、治疗效果预测及治疗方案辅助决策中的应用。
本发明提出了一种基于高通量测序数据和临床表型构建复杂疾病状态评估模型方法在结直肠肿瘤状态评估模型中的应用(包括:结直肠肿瘤状态评估模型构建方法;结直肠肿瘤状态评估panel设计方法;结直肠肿瘤状态评估方法、等),包括基因对应dna突变及rna表达信息,适用于预后、化疗、靶向和免疫治疗的状态评估,所述应用包括以下步骤:
步骤14.1)获取结直肠肿瘤病例信息,包括高通量测序数据和临床信息,根据结直肠肿瘤病例状态分类并进行配对整理,并确定挖掘模式;
步骤14.2)构建结直肠肿瘤相关的基因异常调控关系和基因变异标志物组合;
步骤14.3)筛选结直肠肿瘤相关的临床信息及检验和病理指标;参考结直肠肿瘤相关的基因异常调控关系和步骤14.2所得基因变异标志物组合,整合优化为结直肠肿瘤多元标志物组合,用于构建结直肠肿瘤综合状态评分模型,并开发封装为结直肠肿瘤综合状态评分计算系统;
步骤14.4)基于步骤14.2所得结直肠肿瘤相关的基因异常调控关系和基因变异标志物组合,设计结直肠肿瘤综合状态评估的靶基因目标区域相关探针和/或引物,用作结直肠肿瘤综合状态评估基因检测panel;
步骤14.5)构建一套结直肠肿瘤综合状态评估基因检测panel和综合状态评分计算系统联用流程,使得用户根据需求依此流程可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤14.1中,对结直肠肿瘤病例信息进行分类整理:
步骤14.1.1)将所述结直肠肿瘤病例信息分为转录组数据、外显子组/基因组数据和临床信息;
步骤14.1.2)将所述结直肠肿瘤病例信息按照疾病状态分类并进行配对整理。
具体地,步骤14.2中,构建结直肠肿瘤标志物组合,使用基于贪婪算法的逐次迭代和/或基于遗传算法的进化迭代进行组合优化筛选:
若所述结直肠肿瘤病例信息仅涉及转录组数据与临床信息,则执行步骤14.2.1)基于转录组数据与临床信息进行标志物挖掘,构建结直肠肿瘤相关的基因异常调控关系标志物组合;
若所述结直肠肿瘤病例信息仅涉及外显子组/基因组数据与临床信息,则执行步骤14.2.2)基于外显子组/基因组数据与临床信息进行标志物挖掘,构建结直肠肿瘤相关的基因变异标志物组合;
若所述结直肠肿瘤病例信息同时包含转录组数据、外显子组/基因组数据与临床信息,则执行步骤14.2.3)基于转录组数据、外显子组/基因组数据与临床信息进行标志物挖掘,构建结直肠肿瘤相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤14.2.1)具体包括以下子步骤:
步骤14.2.1.1)构建参考基因调控网络;
步骤14.2.1.2)基于结直肠肿瘤特定疾病状态下的转录组数据以及所述参考基因调控网络的tf-target关系,构建条件特异的基因调控网络;
步骤14.2.1.3)量化条件特异的基因调控网络中的基因调控强度和网络间调控强度差异;
步骤14.2.1.4)筛选不同结直肠肿瘤疾病状态下的条件特异的基因调控网络之间的基因异常调控关系;
步骤14.2.1.5)基于步骤14.2.1.4)得到的基因异常调控关系,构建结直肠肿瘤相关的基因异常调控关系标志物组合。
具体地,步骤14.2.1.2)中,采用基于机器学习的特征选择算法,包括boruta、
具体地,步骤14.2.1.3)中,采用多元线性回归模型量化条件特异的基因调控网络中的基因调控强度;
采用de-biasedlasso方法进行回归,求解得到每一个基因调控关系的调控强度及其置信区间,通过比较不同条件特异基因调控网络中同一调控关系的置信区间是否有重叠,判定其调控差异是否显著;或通过比较不同条件特异基因调控网络中同一调控关系的强度均值变化,无需计算置信区间,直接量化其调控差异。
具体地,步骤14.2.1.4)中,整合三方面与基因调控相关的因素,筛选结直肠肿瘤不同疾病状态下的条件特异基因调控网络之间的基因异常调控关系,包括:基因调控强度显著变化、调控目标基因表达水平显著变化,以及tf对target的调控强度变化方向与target表达水平变化方向一致;同时,根据调控强度在不同疾病状态间的差异程度,对筛选到的基因异常调控关系进行排序。
具体地,步骤14.2.1.5)中以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建结直肠肿瘤相关的基因异常调控关系标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤14.2.2)具体包括以下子步骤:
步骤14.2.2.1)识别与结直肠肿瘤相关的基因变异;
步骤14.2.2.2)采用数据驱动和/或先验知识驱动定量筛选结直肠肿瘤状态相关的重要基因变异;
步骤14.2.2.3)基于步骤14.2.2.2)得到的结直肠肿瘤状态相关的重要基因变异,构建结直肠肿瘤相关的基因变异标志物组合。
具体地,步骤14.2.2.2)中,数据定量过滤筛选,涉及体细胞基因变异频率计算、排序,以及高频变异基因识别,其中基因变异频率≥5%的基因进一步用于先验知识过滤;先验知识过滤筛选,包括应用标准、临床治疗指南、药物标签及通用知识库和文献报道中的结直肠肿瘤相关基因。
具体地,步骤14.2.2.3)中,以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建结直肠肿瘤相关的基因变异标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤14.2.3)具体包括以下子步骤:
步骤14.2.3.1)对于同时具备转录组数据和外显子组/基因组数据的结直肠肿瘤数据集,利用步骤14.2.1.1~14.2.1.4筛选疾病状态相关的基因异常调控关系,同时利用步骤14.2.2.1~14.2.2.2挖掘疾病状态相关的重要基因变异,分别得到结直肠肿瘤相关的基因异常调控关系和重要基因变异;
步骤14.2.3.2)随后采纳步骤14.2.1.5和步骤14.2.2.3中,基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,整合rna和dna信息,构建结直肠肿瘤相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤14.3中,筛选结直肠肿瘤相关的临床信息及检验和病理指标包括以下步骤:
步骤14.3.1)针对已知先验知识,筛选结直肠肿瘤状态相关的临床信息及检验和病理指标;
步骤14.3.2)从结直肠肿瘤队列中病例信息出发,筛选结直肠肿瘤状态相关的临床信息及检验和病理指标。
具体地,所述步骤14.3中,结直肠肿瘤相关基因异常调控关系通过以下方法得到:
将得到的结直肠肿瘤相关的基因异常调控关系和/或基因变异标志物组合,同步骤14.3.1和14.3.2筛选所得结直肠肿瘤状态相关的临床信息及检验和病理指标整合,优化为结直肠肿瘤多元标志物组合。
具体地,所述步骤14.4中,基因检测panel设计包括以下步骤:
步骤14.4.1)基于筛选得到结直肠肿瘤相关基因异常调控关系和/或基因变异标志物组合,并最终纳入结直肠肿瘤综合状态评分方法的基因集,梳理基因集中基因相关信息,去除冗余,确定标准基因名;
步骤14.4.2)针对步骤14.4.1)中梳理后的基因,选择用于结直肠肿瘤检测设计的靶基因目标区域,可用于探针设计或引物设计;
步骤14.4.3)根据步骤14.4.2)中的靶基因目标区域,设计相应的探针和/或引物序列,并记录重要注释;
步骤14.4.4)针对步骤14.4.2)中的靶基因目标区域,参考人类基因组中可设计探针和/或引物数据集,对靶基因目标区域进行优化设计,使探针和/或引物能均匀捕获覆盖目标区域;
步骤14.4.5)将步骤14.4.3和14.4.4中的靶基因目标区域相关探针和/或引物设计区域进行比对,获取具有最优覆盖度的靶基因目标区域相关探针和/或引物设计方案;
步骤14.4.6)基于步骤14.4.5设计的靶基因目标区域相关探针和/或引物,制作出用于充分进行结直肠肿瘤状态评估的基因检测panel。
具体地,步骤14.5中,所述联用流程包括以下步骤:
步骤14.5.1)基于本发明所述方法设计的基因检测panel,得到结直肠肿瘤相关基因异常调控关系和/或基因变异标志物组合的定量值,输入结直肠肿瘤综合状态评分计算系统;
步骤14.5.2)将获取的结直肠肿瘤状态相关的临床信息及检验和病理指标的定量值,输入结直肠肿瘤综合状态评分计算系统;
步骤14.5.3)将步骤14.5.1)和14.5.2)所涉及的硬件、软件和/或在线工具,组合为一套配套联用的流程,使得用户根据需求可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤14.2)中结直肠肿瘤相关基因异常调控关系和基因变异标志物组合,具体基因集包括以下53个基因:runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1、kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e之任一及其组合;具体地,所有53个基因组合可用于生存预后评估;runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1用于化疗方案效果预测;kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1用于靶向治疗方案效果预测,pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e用于结直肠肿瘤免疫浸润与免疫细胞毒性状态评估,以及免疫检查点抑制剂治疗效果预测。
步骤14.3中结直肠肿瘤相关的临床信息及检验和病理指标,同结直肠肿瘤相关基因异常调控关系和基因变异标志物组合的53个基因,一起组成结直肠肿瘤多元标志物组合,用于预后效果、化疗、靶向治疗和免疫治疗效果预测,辅助临床决策;具体地,所有53个基因均用于生存预后评估,其低评分组预示病例预后效果较好;其中runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1用于化疗方案效果预测(特别是术后场景),包括5-fu和联合adjc(包括folfiri、folfox和fufol),为基于病理分期的半定量化疗方案选择,提供定量评分,低评分组病例更能从化疗中受益;kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1用于靶向治疗方案效果预测,其相应基因表达或变异评分,同靶向药受益密切相关,如her2高评分病例,更可能从her2单抗药物治疗获益;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e用于结直肠肿瘤免疫浸润与免疫细胞毒性状态评估,以上基因评分的免疫低风险亚型,免疫细胞浸润程度高,免疫细胞毒性强,免疫检查点激活程度高,更易从免疫检查点抑制剂治疗中受益。
具体地,步骤14.4中设计的结直肠肿瘤综合状态评估的53个靶基因目标区域相关探针和/或引物,对靶基因目标区域覆盖度不低于95%,对其中重要基因变异位点的覆盖度不低于97%;以上53个靶基因目标区域,既可整体作为一个检测panel,也根据具体用途分为3个检测panel,包括化疗状态评估检测panel(包括runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1等基因,),靶向治疗状态评估检测panel(包括ras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1等基因)和免疫治疗状态评估检测panel(包括pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e)。
本发明步骤14.1所述数据获取整理,充分涵盖已公开结直肠肿瘤数据集,包括但不限于tcga、geo、icgc等,并纳入生存、用药治疗效果等信息,并实现对以上信息相关转录组和外显子组标志物的系统挖掘。
本发明步骤14.2所述方法,整合三方面与基因调控相关的因素,筛选结直肠肿瘤cgrn之间的基因异常调控关系,包括:tf-target调控强度显著变化、target表达水平显著变化以及tf对target的调控强度变化方向与target表达水平变化方向一致。同时,可根据调控强度的差异程度,对筛选到的基因异常调控关系进行排序;并基于对病例预后生存和治疗方案效果预测能力,采用基于贪婪算法的逐次增加迭代,挖掘转录组相关标志物,且该标志物组合具有准确可靠,机制可解释性强的特点。
本发明步骤14.2所述方法,综合采用数据驱动和先验知识驱动的定量筛选策略,使用了基于遗传算法的进化迭代方法,筛选结直肠肿瘤状态如进展阶段、预后生存、治疗方案敏感性相关的高频dna变异标志物组合,且该标志物组合具有准确可靠,机制可解释性强的特点。
本发明步骤14.3所述基因集和模型系统,可以实现结直肠癌病人的综合状态评分,且该评分同结直肠肿瘤预后生存和治疗手段(包括但不限于化疗、靶向、免疫抑制剂等)效果有较高相关性。具体来看,所有输入特征均对生存预后有所贡献;但对治疗手段效果预测的权重有所不同,其中runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1的贡献集中在化疗方案效果预测,包括5-fu和联合adjc(包括folfiri、folfox和fufol),为基于病理分期的半定量化疗方案选择,提供有效的定量评分支持;kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1的贡献侧重于靶向治疗方案效果预测,而pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e侧重于结直肠肿瘤免疫浸润与免疫细胞毒性状态评估,对免疫抑制剂治疗方案效果预测贡献较大;手术情况(有/无)、病理级别(i-iv)和微卫星不稳定(msi)等信息对于预后和治疗效果预测均有贡献。
本发明步骤14.4和14.5所述panel设计及评估系统联用流程,可实现探针设计捕获效率和目标区域覆盖度均较高,panel和评分模块可以根据需求灵活调整,用于结直肠肿瘤病人综合状态评估,并辅助临床决策包括但不限于辅助手术方案、化疗方案和靶向治疗方案选择、免疫疗法参考、预后状态评估等。panel和评分模块的灵活调整,示例如下:仅使用异常调控4-dysreg涵盖的7个基因(runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1)标志物组合,可以作为一个小panel,并保留相关的状态评分模型即可组成一个定位于结直肠癌辅助化疗方案状态评估流程。以上思路,同样适用于靶向治疗和免疫抑制剂治疗方案状态评估流程的单独提取,缩小panel,降低检测成本。
本发明提出了一种基于高通量测序数据和临床表型构建复杂疾病状态评估方法在胰腺导管癌状态评估中的应用,包括以下步骤:
步骤15.1)获取胰腺导管癌病例信息,包括高通量测序数据和临床信息,根据胰腺导管癌病例状态分类并进行配对整理;
步骤15.2)构建胰腺导管癌相关的基因异常调控关系和基因变异标志物组合;
步骤15.3)筛选胰腺导管癌相关的临床信息及检验和病理指标;参考胰腺导管癌相关的基因异常调控关系和同步骤15.2所得胰腺导管癌相关的基因异常调控关系和基因变异标志物组合,整合优化为胰腺导管癌多元标志物组合,用于构建胰腺导管癌综合状态评分模型,并开发封装为胰腺导管癌综合状态评分计算系统;
步骤15.4)基于步骤15.2所得胰腺导管癌相关基因异常调控关系和基因变异标志物组合,设计胰腺导管癌综合状态评估的靶基因目标区域相关探针和/或引物,用作胰腺导管癌综合状态评估基因检测panel;
步骤15.5)构建一套胰腺导管癌综合状态评估基因检测panel和综合状态评分计算系统联用流程,使得用户根据需求依此流程可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤15.1中,对胰腺导管癌病例信息进行分类整理:
步骤15.1.1)将所述胰腺导管癌病例信息分为转录组数据、外显子组/基因组数据和临床信息;
步骤15.1.2)将所述胰腺导管癌病例信息按照疾病状态分类并进行配对整理。
具体地,步骤15.2中,构建胰腺导管癌标志物组合,使用基于贪婪算法的逐次迭代和/或基于遗传算法的进化迭代进行组合优化筛选:
若所述胰腺导管癌病例信息仅涉及转录组数据与临床信息,则执行步骤15.2.1)基于转录组数据与临床信息进行标志物挖掘,构建胰腺导管癌相关的基因异常调控关系标志物组合;
若所述胰腺导管癌病例信息仅涉及外显子组/基因组数据与临床信息,则执行步骤15.2.2)基于外显子组/基因组数据与临床信息进行标志物挖掘,构建胰腺导管癌相关基因变异标志物组合;
若所述胰腺导管癌病例信息同时包含转录组数据、外显子组/基因组数据与临床信息,则执行步骤15.2.3)基于转录组数据、外显子组/基因组数据与临床信息进行标志物挖掘,构建胰腺导管癌相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤15.2.1)具体包括以下子步骤:
步骤15.2.1.1)构建参考基因调控网络;
步骤15.2.1.2)基于胰腺导管癌特定疾病状态下的转录组数据以及所述参考基因调控网络的tf-target关系,构建条件特异的基因调控网络;
步骤15.2.1.3)量化条件特异的基因调控网络中的基因调控强度和网络间调控强度差异;
步骤15.2.1.4)筛选胰腺导管癌不同疾病状态下的条件特异的基因调控网络之间的基因异常调控关系;
步骤15.2.1.5)基于步骤15.2.1.4)得到的基因异常调控关系,构建胰腺导管癌相关的基因异常调控关系标志物组合。
具体地,步骤15.2.1.2)中,采用基于机器学习的特征选择算法,包括boruta、
具体地,步骤15.2.1.3)中,采用多元线性回归模型量化条件特异基因调控网络中的基因调控强度;
采用de-biasedlasso方法进行回归,求解得到每一个基因调控关系的调控强度及其置信区间,通过比较不同条件特异基因调控网络中同一调控关系的置信区间是否有重叠,判定其调控差异是否显著;或通过比较不同条件特异基因调控网络中同一调控关系的强度均值变化,无需计算置信区间,直接量化其调控差异。
具体地,步骤15.2.1.4)中,整合三方面与基因调控相关的因素,筛选胰腺导管癌不同疾病状态下的条件特异基因调控网络之间的基因异常调控关系,包括:基因调控强度显著变化、调控目标基因表达水平显著变化以及tf对target的调控强度变化方向与target表达水平变化方向一致;同时,根据调控强度在不同疾病状态间的差异程度,对筛选到的基因异常调控关系进行排序。
具体地,步骤15.2.1.5)中以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建胰腺导管癌相关基因异常调控关系标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤15.2.2)具体包括以下子步骤:
步骤15.2.2.1)识别与胰腺导管癌相关的基因变异;
步骤15.2.2.2)采用数据驱动和/或先验知识驱动定量筛选胰腺导管癌状态相关的重要基因变异;
步骤15.2.2.3)基于步骤15.2.2.2)得到的胰腺导管癌状态相关的重要基因变异,构建胰腺导管癌相关的基因变异标志物组合。
具体地,步骤15.2.2.2)中,数据定量过滤筛选,涉及体细胞基因变异频率计算、排序,以及高频变异基因识别,其中基因变异频率≥5%的基因进一步用于先验知识过滤;先验知识过滤筛选,包括应用标准、临床治疗指南、药物标签及通用知识库和文献报道中的胰腺导管癌相关基因。
具体地,步骤15.2.2.3)中,以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建胰腺导管癌相关的基因变异标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤15.2.3)具体包括以下子步骤:
步骤15.2.3.1)对于同时具备转录组数据和外显子组/基因组数据的胰腺导管癌数据集,利用步骤15.2.1.1~15.2.1.4筛选疾病状态相关的基因异常调控关系,同时利用步骤15.2.2.1~15.2.2.2挖掘疾病状态相关的重要基因变异,分别得到胰腺导管癌相关的基因异常调控关系和重要基因变异;
步骤15.2.3.2)随后采纳步骤15.2.1.5和步骤15.2.2.3中,基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,整合rna和dna信息,构建胰腺导管癌相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤15.3中,筛选胰腺导管癌相关的临床信息及检验和病理指标包括以下步骤:
步骤15.3.1)针对已知先验知识,筛选胰腺导管癌状态相关的临床信息及检验和病理指标;
步骤15.3.2)从胰腺导管癌队列中病例信息出发,筛选胰腺导管癌状态相关的临床信息及检验和病理指标。
具体地,所述步骤15.3中,胰腺导管癌相关基因异常调控关系通过以下方法得到:
将得到的胰腺导管癌相关基因异常调控关系和/或基因变异标志物组合,同步骤15.3.1和15.3.2筛选所得胰腺导管癌状态相关的临床信息及检验和病理指标整合,优化为胰腺导管癌多元标志物组合。
具体地,所述步骤15.4中,基因检测panel设计包括以下步骤:
步骤15.4.1)基于筛选得到胰腺导管癌相关基因异常调控关系和/或基因变异标志物组合,并最终纳入胰腺导管癌综合状态评分方法的基因集,梳理基因集中基因相关信息,去除冗余,确定标准基因名;
步骤15.4.2)针对步骤15.4.1)中梳理后的基因,选择用于胰腺导管癌检测设计的靶基因目标区域,可用于探针设计或引物设计;
步骤15.4.3)根据步骤15.4.2)中的靶基因目标区域,设计相应的探针和/或引物序列,并记录重要注释;
步骤15.4.4)针对步骤15.4.2)中的靶基因目标区域,参考人类基因组中可设计探针和/或引物数据集,对靶基因目标区域进行优化设计,使探针和/或引物能均匀捕获覆盖目标区域;
步骤15.4.5)将步骤15.4.3和15.4.4中的靶基因目标区域相关探针和/或引物设计区域进行比对,获取具有最优覆盖度的靶基因目标区域相关探针和/或引物设计方案;
步骤15.4.6)基于步骤15.4.5设计的靶基因目标区域相关探针和/或引物,制作出用于充分进行胰腺导管癌状态评估的基因检测panel。
具体地,步骤15.5中,所述联用流程包括以下步骤:
步骤15.5.1)基于本发明所述方法设计的基因检测panel,得到胰腺导管癌相关基因异常调控关系和/或基因变异标志物组合的定量值,输入胰腺导管癌综合状态评分计算系统;
步骤15.5.2)将获取的胰腺导管癌状态相关的临床信息及检验和病理指标的定量值,输入胰腺导管癌综合状态评分计算系统;
步骤15.5.3)将步骤15.5.1)和15.5.2)所涉及的硬件、软件和/或在线工具,组合为一套配套联用的流程,使得用户根据需求可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤15.2中胰腺导管癌相关基因异常调控关系和基因变异标志物组合,具体基因集包括以下86个基因:akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、tp53、tsc1、tsc2、smarcb1、smad4、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e之任一或其组合。具体的,所有86个基因组合可用于生存预后评估;kras/tp53/cdkn2a及所有基因拷贝数变异用于手术方案效果预测;所有基因拷贝数变异用于化疗方案效果预测;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e用于胰腺导管癌病人的免疫浸润与免疫细胞毒性状态评估,以及免疫检查点抑制剂治疗效果预测;akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、tp53、tsc1、tsc2、smarcb1、smad4、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1、stk11用于潜在靶向治疗效果预测。
具体地,步骤15.3中胰腺导管癌相关的临床信息及检验和病理指标,主要包括胰腺导管癌病人的年龄、性别、血液生化及免疫检测指标、手术情况(有/无)、病理级别(i-iv)和肿瘤病人移植动物模型(pdx)建模情况(快/慢/无)等临床信息,同本发明所述胰腺导管癌相关基因异常调控关系和基因变异标志物组合的86个基因,一起组成胰腺导管癌多元标志物组合,用于预后效果、化疗、免疫治疗和潜在靶向治疗效果预测,辅助临床决策。具体的,所有86个基因组合可用于生存预后评估,其低评分组预示病例预后效果较好;kras/tp53/cdkn2a及所有基因拷贝数变异用于手术方案效果预测,其中低风险分类病例更易从r0范式手术治疗受益;所有86个基因拷贝数变异用于化疗方案效果预测,拷贝变异评分越高的病例越易从吉西他滨(gemcitabine)治疗受益,拷贝变异评分越低的病例越易从伊立替康(irinotecan)治疗受益;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e用于胰腺导管癌病人的免疫浸润与免疫细胞毒性状态评估,以及免疫检查点抑制剂治疗效果预测,以上基因评分的免疫低风险亚型病例,免疫细胞浸润程度高,免疫细胞毒性强,免疫检查点激活程度高,更易从免疫检查点抑制剂治疗中受益;akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、tp53、tsc1、tsc2、smarcb1、smad4、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1、stk11用于潜在靶向治疗效果预测,其相应基因表达或变异评分,同靶向药受益密切相关,如kras、nras、akt、brca2等突变病例,预测更难以从抗egfr单抗治疗中获益。肿瘤病人移植动物模型(pdx)建模情况,可用于手术方案效果预测,造模不成功病例更易从手术中受益。
具体地,步骤15.4中设计的胰腺导管癌综合状态评估的86个靶基因目标区域相关探针和/或引物,对靶基因目标区域覆盖度不低于95%,对其中重要基因变异位点的覆盖度不低于97%;以上86个靶基因目标区域,既可整体作为一个检测panel(用于预后状态评估和化疗状态评估预测),也根据具体用途分为3个检测panel,包括手术状态评估检测panel(kras/tp53/cdkn2a及所有基因拷贝数变异),免疫治疗状态评估检测panel(pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e),潜在靶向治疗状态评估检测panel(akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、tp53、tsc1、tsc2、smarcb1、smad4、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1、stk11)。
本发明中,胰腺导管癌病人的年龄、性别、病理级别、血液生化及免疫指标(如ca199血清浓度等)、手术情况r0-r2、pdx建模情况作为补充临床信息,也可纳入评分模型的输入范围。
本发明中,步骤15.2综合采用数据驱动和先验知识驱动的定量筛选策略,筛选胰腺导管状态如进展阶段、预后生存、治疗方案敏感性相关的高频dna变异标志物组合,可包括基因变异、临床病理和pdx建模数据等信息,且该标志物组合具有准确可靠,机制可解释性强的特点。同时在标志物组合优化阶段,根据需要,灵活采用基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,提升效果。
本发明中,步骤15.3所述基因集和模型系统,可以实现胰腺导管癌病人的综合状态评分,且该评分同胰腺导管癌病人的预后生存和治疗手段(包括但不限于手术范式、化疗、靶向、免疫抑制剂等)效果有较高相关性。所有输入特征均对生存预后有所贡献;但对治疗手段效果预测的权重有所不同,其中kras/tp53/cdkn2a及所有基因拷贝数变异的贡献集中在手术方案效果预测;所有基因拷贝数变异的贡献侧重于化疗方案效果预测,特别是吉西他滨(gemcitabine)和伊立替康(irinotecan)的疗效预测;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e侧重于胰腺导管癌病人的免疫浸润与免疫细胞毒性状态评估,对免疫抑制剂治疗方案效果预测贡献较大;另外,对于部分处于临床试验中的可能用于胰腺导管癌治疗的靶向药,akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、tp53、tsc1、tsc2、smarcb1、smad4、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1、stk11的突变情况可以提供有价值的参考。不仅胰腺导管癌病人的年龄、性别、病理级别、血液生化及免疫指标(如ca199血清浓度等)、手术情况r0-r2等临床信息,而且病例的pdx建模情况,对于病例的预后效果预测均有贡献。
本发明中,步骤15.4和15.5所述panel设计及评估系统联用流程,可实现探针设计捕获效率和目标区域覆盖度均较高,panel和评分模块可以根据需求灵活调整,用于胰腺导管癌病人综合状态评估,并辅助临床决策包括但不限于手术方案、辅助化疗方案和靶向治疗方案选择、免疫疗法参考、预后状态评估等。panel和评分模块的灵活调整示例如下,选取43个基因,包括akt1、brca2、erbb2、idh1、map2k2、mtor、pms1、apc、cdkn2a、fbxw7、jak2、met、nras、pms2、ar、cftr、fgfr1、fgfr2、fgfr3、kit、mlh1、ntrk1、pten、braf、ctnnb1、kras、msh2、msh6、pik3ca、pik3r1、ret、ros1、brca1、egfr、map2k1、smarca4、stk11、tp53、tsc1、tsc2、smarcb1、smad4等组成一个小panel并保留相关的状态评分模型,即可形成一个胰腺导管癌病例手术和辅助化疗方案状态评估流程。以上思路,同样适用于胰腺导管癌预后和免疫抑制剂治疗方案等状态评估流程的单独提取构建,缩小panel,降低检测成本。
本发明提出了一种基于高通量测序数据和临床表型构建复杂疾病状态评估方法在泛肿瘤靶向药敏感性状态评估中的应用,包括以下步骤:
步骤16.1)获取泛肿瘤癌病例信息,包括高通量测序数据和临床信息,根据泛肿瘤病例状态分类并进行配对整理,并确定挖掘模式;
步骤16.2)构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的基因异常调控关系标志物组合;
步骤16.3)筛选泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的临床信息及检验和病理指标;参考泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的基因异常调控关系和步骤16.2所得泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的基因异常调控关系标志物组合,整合优化为泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关多元标志物组合,用于构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性综合状态评分模型,并开发封装为泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性综合状态评分计算系统;
步骤16.4)基于步骤16.2所得泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因异常调控关系标志物组合,设计泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性综合状态评估的靶基因目标区域相关探针和/或引物,用作泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性综合状态评估基因检测panel。
步骤16.5)构建一套泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性综合状态评估基因检测panel和综合状态评分计算系统联用流程,使得用户根据需求依此流程可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤16.1中,对泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息进行分类整理:
步骤16.1.1)将所述泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息分为转录组数据、外显子组/基因组数据和临床信息;
步骤16.1.2)将所述泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息按照疾病状态分类并进行配对整理。
具体地,步骤16.2中,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性标志物组合,使用基于贪婪算法的逐次迭代和/或基于遗传算法的进化迭代进行组合优化筛选:
若所述泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息仅涉及转录组数据与临床信息,则执行步骤16.2.1)基于转录组数据与临床信息进行标志物挖掘,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因异常调控关系标志物组合;
若所述泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息仅涉及外显子组/基因组数据与临床信息,则执行步骤16.2.2)基于外显子组/基因组数据与临床信息进行标志物挖掘,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因变异标志物组合;
若所述泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性病例信息同时包含转录组数据、外显子组/基因组数据与临床信息,则执行步骤16.2.3)基于转录组数据、外显子组/基因组数据与临床信息进行标志物挖掘,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因异常调控关系和基因变异标志物组合。
具体地,所述步骤16.2.1)具体包括以下子步骤:
步骤16.2.1.1)构建参考基因调控网络;
步骤16.2.1.2)基于特定疾病状态下的转录组数据以及所述参考基因调控网络的tf-target关系,构建条件特异的基因调控网络;
步骤16.2.1.3)量化条件特异的基因调控网络中的基因调控强度和网络间调控强度差异;
步骤16.2.1.4)筛选不同疾病状态下的条件特异的基因调控网络之间的基因异常调控关系;
步骤16.2.1.5)基于步骤16.2.1.4)得到的基因异常调控关系,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的基因异常调控关系标志物组合。
具体地,步骤16.2.1.2)中,采用基于机器学习的特征选择算法,包括boruta、
具体地,步骤16.2.1.3)中,采用多元线性回归模型量化条件特异基因调控网络中的基因调控强度;
采用de-biasedlasso方法进行回归,求解得到每一个基因调控关系的调控强度及其置信区间,通过比较不同条件特异基因调控网络中同一调控关系的置信区间是否有重叠,判定其调控差异是否显著;或通过比较不同条件特异基因调控网络中同一调控关系的强度均值变化,无需计算置信区间,直接量化其调控差异。
具体地,步骤16.2.1.4)中,整合三方面与基因调控相关的因素,筛选不同疾病状态下的条件特异基因调控网络之间的基因异常调控关系,包括:基因调控强度显著变化、调控目标基因表达水平显著变化以及tf对target的调控强度变化方向与target表达水平变化方向一致;同时,根据调控强度在不同疾病状态间的差异程度,对筛选到的基因异常调控关系进行排序。
具体地,步骤16.2.1.5)中以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因异常调控关系标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤16.2.2)具体包括以下子步骤:
步骤16.2.2.1)识别与泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关的基因变异;
步骤16.2.2.2)采用数据驱动和/或先验知识驱动定量筛选泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性状态相关的重要基因变异;
步骤16.2.2.3)基于步骤16.2.2.2)得到的泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性状态相关的重要基因变异,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因变异标志物组合。
具体地,步骤16.2.2.2)中,数据定量过滤筛选,涉及体细胞基因变异频率计算、排序,以及高频变异基因识别,其中基因变异频率≥5%的基因进一步用于先验知识过滤;先验知识过滤筛选,包括应用标准、临床治疗指南、药物标签及通用知识库和文献报道中的泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因。
具体地,步骤16.2.2.3)中,以基于贪婪算法的逐次增加迭代,和/或基于遗传算法的进化迭代,构建泛肿瘤靶向药敏感性泛肿瘤靶向药敏感性相关基因变异标志物组合;对上述标志物组合,以c-index为指标衡量其对疾病预后状态的预测效果,或以auc为指标衡量其对治疗方案受益状态的预测效果。
具体地,所述步骤16.2.3)具体包括以下子步骤:
步骤16.2.3.1)对于同时具备转录组数据和外显子组/基因组数据的泛肿瘤靶向药敏感性数据集,利用步骤16.2.1.1~16.2.1.4筛选疾病状态相关的基因异常调控关系,同时利用步骤16.2.2.1~16.2.2.2挖掘疾病状态相关的重要基因变异,分别得到泛肿瘤靶向药敏感性相关的基因异常调控关系和重要基因变异;
步骤16.2.3.2)随后采纳步骤16.2.1.5和步骤16.2.2.3中,基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,整合rna和dna信息,构建泛肿瘤靶向药敏感性相关的基因异常调控关系和基因变异标志物组合。
具体地,所述步骤16.3中,筛选泛肿瘤靶向药敏感性相关的临床信息及检验和病理指标包括以下步骤:
步骤16.3.1)针对已知先验知识,筛选泛肿瘤靶向药敏感性状态相关的临床信息及检验和病理指标;
步骤16.3.2)从泛肿瘤靶向药敏感性队列中病例信息出发,筛选泛肿瘤靶向药敏感性状态相关的临床信息及检验和病理指标。
具体地,所述步骤16.3中,泛肿瘤靶向药敏感性相关基因异常调控关系通过以下方法得到:
将得到的泛肿瘤靶向药敏感性相关基因异常调控关系和/或基因变异标志物组合,同步骤16.3.1和16.3.2筛选所得泛肿瘤靶向药敏感性状态相关的临床信息及检验和病理指标整合,优化为泛肿瘤靶向药敏感性多元标志物组合。
具体地,所述步骤16.4中,基因检测panel设计包括以下步骤:
步骤16.4.1)基于筛选得到泛肿瘤靶向药敏感性相关基因异常调控关系和/或基因变异标志物组合,并最终纳入泛肿瘤靶向药敏感性综合状态评分方法的基因集,梳理基因集中基因相关信息,去除冗余,确定标准基因名;
步骤16.4.2)针对步骤16.4.1)中梳理后的基因,选择用于泛肿瘤靶向药敏感性检测设计的靶基因目标区域,可用于探针设计或引物设计;
步骤16.4.3)根据步骤16.4.2)中的靶基因目标区域,设计相应的探针和/或引物序列,并记录重要注释;
步骤16.4.4)针对步骤16.4.2)中的靶基因目标区域,参考人类基因组中可设计探针和/或引物数据集,对靶基因目标区域进行优化设计,使探针和/或引物能均匀捕获覆盖目标区域;
步骤16.4.5)将步骤16.4.3和16.4.4中的靶基因目标区域相关探针和/或引物设计区域进行比对,获取具有最优覆盖度的靶基因目标区域相关探针和/或引物设计方案;
步骤16.4.6)基于步骤16.4.5设计的靶基因目标区域相关探针和/或引物,制作出用于充分进行泛肿瘤靶向药敏感性状态评估的基因检测panel。
具体地,步骤16.5中,所述联用流程包括以下步骤:
步骤16.5.1)基于本发明所述方法设计的基因检测panel,得到泛肿瘤靶向药敏感性相关基因异常调控关系和/或基因变异标志物组合的定量值,输入泛肿瘤靶向药敏感性综合状态评分计算系统;
步骤16.5.2)将获取的泛肿瘤靶向药敏感性状态相关的临床信息及检验和病理指标的定量值,输入泛肿瘤靶向药敏感性综合状态评分计算系统;
步骤16.5.3)将步骤16.5.1)和16.5.2)所涉及的硬件、软件和/或在线工具,组合为一套配套联用的流程,使得用户根据需求可以完成检测、信息输入、计算评估和结果获取。
具体地,步骤16.2中泛肿瘤靶向药敏感性相关基因异常调控关系标志物组合,特别适用于tgfbeta通路、mapk通路和pi3k通路相关的11种靶向用药治疗方案,包括binimetinib、bkm120、byl719、byl719+cetuximab、byl719+cetuximab+encorafenib、byl719+encorafenib、byl719+ljm716、cetuximab、cetuximab+encorafenib、clr457、encorafenib,具体基因集包括以下24个基因:axin1、junb、myc、smad5、smad4、tgif2、ubb、atf3、bmpr2、jund、klf10、nr2c2、ppp1cb、skil、smurf1、sp1、tp53、pitx2、tfdp2、e2f4、smad1、klf6、smad3、klf11。同时,对于胃肠道肿瘤tgfbeta通路相关靶向用药,bmpr2、myc、tfdp2、tgif2等四个基因可以作为一个基因异常调控关系标志物组合。
具体地,步骤16.3中泛肿瘤靶向药敏感性多元标志物组合构建方法,利用基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,进行多元标志物组合优化,通过机器学习分类算法,决策树、随机森林、svm均可采用,构建泛肿瘤靶向药敏感性综合状态评分模型,并开发封装为泛肿瘤靶向药敏感性综合状态评分计算系统,用于泛肿瘤病例靶向用药效果预测。
具体地,步骤16.3中泛肿瘤病人靶向用药相关的临床信息及检验和病理指标,主要包括肿瘤病人的年龄、性别、血液生化及免疫检测指标、手术情况(有/无)、病理级别(分化程度/tnm分期)、转移和治疗等临床信息,同本发明所述泛肿瘤靶向药敏感性相关基因异常调控关系标志物组合的24个基因,一起组成泛肿瘤靶向药敏感性多元标志物组合,用于泛肿瘤靶向药敏感性治疗效果预测,特别是tgfbeta-mapk-pi3k三通路靶向用药治疗效果预测,辅助临床决策。具体地,可基于泛肿瘤靶向药敏感性多元标志物组合,构建开发泛肿瘤靶向药敏感性综合状态评分计算系统,用于泛肿瘤病例tgfbeta-mapk-pi3k三通路相关6种单药治疗方案(包括binimetinib、bkm120、byl719、cetuximab、clr457和encorafenib)和5种联合治疗方案(包括byl719+cetuximab、byl719+cetuximab+encorafenib、byl719+encorafenib、byl719+ljm716、cetuximab+encorafenib)的治疗受益情况预测,辅助临床决策。
具体地,步骤16.4中设计的泛肿瘤靶向药敏感性状态评估的24个靶基因目标区域相关探针和/或引物,对靶基因目标区域覆盖度不低于95%,对其中重要基因变异位点的覆盖度不低于97%。
本发明泛肿瘤靶向用药方案状态评估方法及应用,步骤16.1所述数据收集整理,充分涵盖已公开泛肿瘤用药数据集,充分利用病人队列和动物实验数据,包括但不限于tcga、geo、nibrpdxe等。
本发明泛肿瘤靶向用药方案状态评估方法及应用,步骤16.2所述方法,整合三方面与基因调控相关的因素,筛选泛肿瘤辅助用药专属cgrn之间的基因异常调控关系,包括:tf-target调控强度显著变化、target表达水平显著变化以及tf对target的调控强度变化方向与target表达水平变化方向一致。同时,可根据调控强度的差异程度,对筛选到的基因异常调控关系进行排序;并基于对收集到的所有用药方案(包括但不限于靶向药单用、靶向药联合用药等)效果预测能力,挖掘转录组相关标志物及组合,且该标志物组合具有准确可靠,机制可解释性强的特点。同时综合采用数据驱动和先验知识驱动的定量筛选策略,并在标志物组合优化阶段,根据需要,灵活采用基于贪婪算法的逐次增加迭代或基于遗传算法的进化迭代,提升效果。
本发明泛肿瘤靶向用药方案状态评估方法及应用,步骤16.3所述方法,可以实现基于生物通路的泛肿瘤靶向药治疗方案效果评估基因集构建,并可实现对泛肿瘤病人的辅助用药治疗综合状态评分,且该评分同泛肿瘤靶向药治疗效果密切相关。此处富集到了tgfbeta通路、mapk通路和pi3k通路的11种靶向用药治疗方案,包括binimetinib、bkm120、byl719、byl719+cetuximab、byl719+cetuximab+encorafenib、byl719+encorafenib、byl719+ljm716、cetuximab、cetuximab+encorafenib、clr457、encorafenib,该评估模型所用基因集,包括24个基因,即包括,axin1、junb、myc、smad5、smad4、tgif2、ubb、atf3、bmpr2、jund、klf10、nr2c2、ppp1cb、skil、smurf1、sp1、tp53、pitx2、tfdp2、e2f4、smad1、klf6、smad3、klf11等。
本发明泛肿瘤靶向用药方案状态评估方法及应用,步骤16.4和16.5所述panel设计及评估系统联用流程,可实现探针设计捕获效率和目标区域覆盖度均较高,panel和评分模块可以根据需求灵活调整,实现对泛肿瘤病人辅助用药治疗综合状态评分,有效辅助临床决策,提升治疗效果。panel和评分模块的灵活调整示例如下,bmpr2、myc、tfdp2、tgif2等4个基因组成的小panel,可以用pcr检测表达量,配合相应评分模型,用于对胃肠道相关肿瘤cetuximab治疗状态评估。以上思路也适用于,针对其它肿瘤类型和用药方案,定制化单独提取特征基因和临床信息,缩小panel,降低检测成本。
本发明的有益效果包括通过基于转录组表达数据,构建条件特异的基因调控网络,能够识别基因异常调控关系;且包含不止一种识别策略;能够由基因异常调控关系构建标志物;且此构建过程包括两种筛选策略,即基于贪婪算法的逐次增加迭代和基于遗传算法的进化迭代,最终构建准确性与机制解释性兼顾的标志物可用于复杂疾病预后评估、治疗效果预测及治疗方案辅助决策等。
本发明的有益效果也包括通过识别复杂疾病相关重要基因变异;且具备不同识别策略,如数据驱动的定量筛选和知识库过滤筛选及其联用;能够由复杂疾病相关dna层面重要变异基因构建标志物;且此构建过程包括两种筛选策略,即基于贪婪算法的逐次增加迭代和基于遗传算法的进化迭代,最终构建的标志物可用于复杂疾病预后评估、治疗效果预测及治疗方案辅助决策等;且可实现rna数据和dna数据的整合利用,方法灵活多样,标志物组合系统,准确性与机制解释性兼备。
本发明的有益效果还包括可利用丰富的技术手段,充分整合高通量测序数据、临床信息和知识库来源的多元信息构建综合评分系统;且包括临床及药物指南和公开文献的系统挖掘检索,临床信息的有效利用,综合评分计算系统构建等策略及功能;同时提供了综合评分计算系统配套的基因检测panel设计方案;且包括基因探针目标区域设计,探针覆盖度设计,基于覆盖度的质量控制;并提出了基因检测panel同综合评分系统的联用流程,且包括综合状态评估模型功能,输入输出功能及可能构思,组合联用形式及可能构思。
本发明的有益效果亦包括提出了一种结直肠肿瘤状态评估模型构建及panel设计方案;包括充分利用多元信息,含dna、rna测序及临床信息等,通过较少的基因和实用可靠的检测评估手段;且实现结直肠肿瘤病例综合状态评估,包括病例预后计算评估,化疗、靶向和免疫治疗等方案治疗效果预测;并辅助临床决策包括但不限于辅助手术方案、化疗方案和靶向治疗方案选择、免疫疗法参考、预后状态评估等。
附图说明
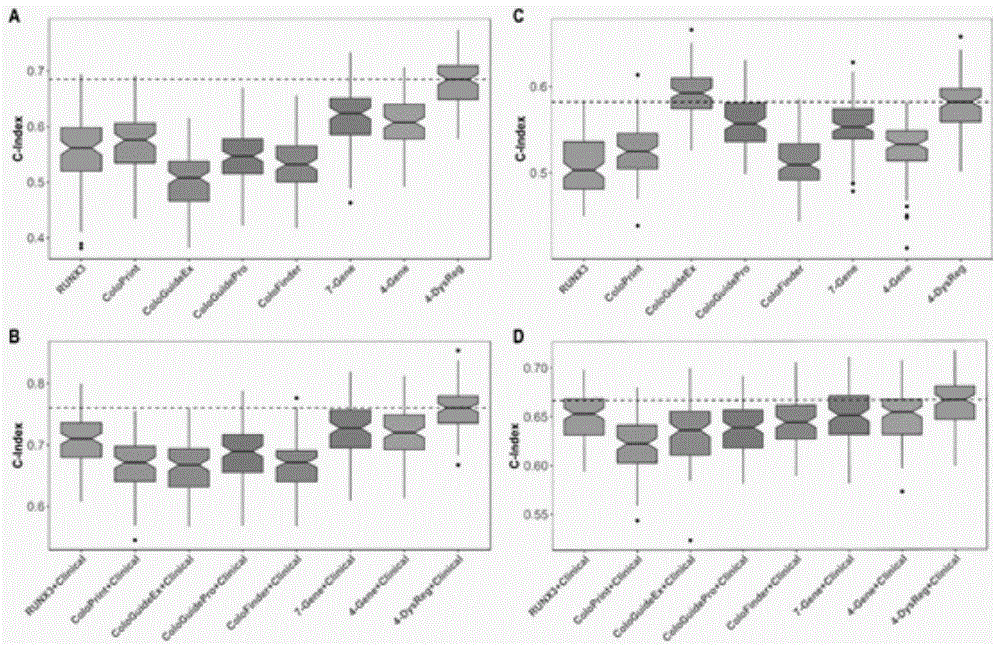
附图1为4-dysreg与其他预后标志物之间的准确性比较。
附图2为结直肠肿瘤综合状态评估流程应用示意图。
具体实施方式
以下结合实施例和附图进一步阐述本发明。应理解,这些实施例仅用于说明本发明,而非限制本发明的范围。在不背离本发明构思的精神和范围下,本领域技术人员能够想到的变化和优点都被包括在本发明中,并且以所附的权利要求书及等同内容为保护范围。
本发明的实施例应用于结直肠肿瘤状态评估模型构建及panel设计的全过程,结合具体实施例对本发明做进一步详细说明,应理解,以下实施例仅用于说明本发明而非用于限定本发明的范围。其具体步骤如下:
s1.1结直肠肿瘤测序数据和临床表型信息的获取与整理
tcga-crc的mrna数据和临床数据从ucscxena数据库下载。380个原位肿瘤样本和51个癌旁样本被挑选出来。mrna数据的表达水平以tpm量化。当tpm的值小于1时,看作是缺失值。对于一个基因,如果缺失值的数量大于样本量的20%,就把该基因去除。剩下的缺失值用k最小近邻法(k-nearestneighbor,knn)进行填充,并做log2转化。在这些样本中,32对配对的肿瘤样本和癌旁样本用于做基因异常调控分析。包括完整的总生存时间(overallsurvival,os)、无进展生存期(recurrentfreesurvival,rfs)、年龄、性别、病理级别的350个原位肿瘤样本用于后续标志物挖掘。
gse39582和gse17538两crc数据集的表达谱数据和临床数据从geo(http://www.ncbi.nlm.nih.gov/geo/)下载。gse39582包含566个肿瘤样本和19个正常的样本,gse17538包含238肿瘤样本,这两个数据集都基于gpl570表达谱芯片平台。对于一个探针能够映射多个基因的情况,将相应的探针从数据集中去除;对于多个探针映射到一个基因的情况,用每个样本中的相应的多个探针的最大值作为该基因在每个样本中的表达值。缺失值的处理方式与tcga-crc中缺失值的处理方式一致。最后利用分位数方法做样本间的标准化,并做log2转化。gse39582数据集中有563个样本拥有完整的如上所述的临床信息,gse17538数据集中有200个样本拥完整的临床信息,这些样本用于后续标志物挖掘。
tcga-crc的体细胞突变数据(somaticmutation)和拷贝数变异(cnv),也由ucscxena数据库下载,其中体细胞突变数据来自varscan2识别,拷贝数变异以log2(copy-number/2)为单位,病例选择同转录组数据取交集,共得到包括完整的总生存时间(overallsurvival,os)、无进展生存期(recurrentfreesurvival,rfs)、年龄、性别、病理级别的350个原位肿瘤样本用于后续标志物挖掘。
因为本实施例同时包括转录组数据、dna测序数据与临床信息,因此执行步骤2.3,分别挖掘转录组和dna变异标志物。
s1.2基于结直肠肿瘤转录组中的基因异常调控关系挖掘生物标志物
s1.2.1构建结直肠肿瘤基因调控网络并识别基因异常调控关系
基于步骤s1中整理到的tcga-crc数据集中,32对配对的肿瘤样本和癌旁样本,参考步骤2.1.1-2.1.4,利用boruta算法分别构建正常条件和癌症条件的grn,正常条件下的grn有30186个调控关系,癌症条件下的grn有15665个调控关系。利用de-biasedlasso方法对调控关系的调控强度进行度量,并获得调控强度的95%置信区间。随后,根据调控强度差异显著、靶基因表达水平差异显著、调控强度变化方向与靶基因表达水平变化方向一致三个因素筛选异常调控关系,最终筛选出389个基因异常调控关系。
s1.2.2由识别到的基因异常调控关系构建标志物
此处采用步骤2.1.5中基于贪婪算法的逐次增加迭代方法,筛选最优标志物组合。具体步骤如下:
首先,在tcga-crc数据集中,对每个异常调控关系结合临床数据(包括年龄、性别、病理级别)对os拟合cox模型,并用c-index量化模型对预后的准确性。其中,异常调控关系runx3-gpr15对应的模型的c-index最大,达到0.763,遂用作迭代起始。
其次,根据步骤2.1.5所述基于贪婪算法的逐次增加迭代方法,一个包括4个异常调控关系的预后标志物(4-dysreg),即runx3-gpr15、runx3-p2ry8、snai3-tlr7、atoh1-siglec1作为最优组合。
再次,基于4个异常调控关系(4-dysreg)中的7个基因的表达数据,结合tcga-crc数据集的年龄、性别、病理分期临床信息,我们在tcga-crc数据集上建立了oscox模型。该模型的c-index为0.79(se=0.038)。
最后,在tcga-crc和gse39582数据集上,将4-dysreg的预测效果同已报道的crc标志物如runx3、coloprint(包含18个基因)、cologuideex(包含13个基因)、cologuidepro(包含7个基因)、colofinder(包含9个基因)、一个7-gene标志物、和一个4-gene标志物等进行比较,结果表明4-dysreg对预后具有更为稳定良好的预测能力。见附图1,4-dysreg与其他预后标志物之间的准确性比较。
s1.2.3结直肠肿瘤辅助化疗状态评估标志物挖掘及模型构建
辅助化疗(adjuvantchemotherapy,adjc)在治疗crc患者的过程中被广泛采用。adjc的指导原则建立在病理级别上。iii期和iv期crc患者常规治疗接受adjc治疗;具有高复发风险的二期crc患者也考虑接受adjc。相对目前以经验为主的半定量adjc指导方案,亟需一套adjc相关肿瘤状态评估方案。
利用gse39582中321个没有接受adjc的样本作为训练集,以这4-dysreg中的7个基因作为自变量拟合一个oscox模型,利用该模型计算232个接受adjc的样本的风险评分。结果显示,风险评分为负的样本具有更好的预后(hr=0.432,95%ci:0.269~0.693)。
4-dysreg对特定化疗类型疗效的预测能力,包括5-fu和联合adjc(包括folfiri、folfox和fufol)。结果显示,风险评分为负的样本在联合adjc(n=84,hr=0.380;95%ci:0.178~0.813)和5-fu(n=79,hr=0.437,95%ci:0.181~1.055)中都有更好的预后。利用timeroc评估标志物对联合adjc预测的准确性,3年和5年总生存期的auc分别达到0.71和0.74,高于病理分期模型的auc0.65和0.69。这些结果表明,该项研究构建的标志物4-dysreg对adjc的治疗效果具有预测能力,不仅对单独的5-fu或联合adjc具有预测能力,而且对所有类型的adjc的整体结果也有预测能力。
s1.3基于结直肠肿瘤dna测序数据中的基因变异挖掘生物标志物
s1.3.1利用步骤2.2.2疾病状态相关dna变异的定量化及筛选(如基因拷贝数与体细胞突变,snp、indel、cnv、fusion等)所述方法,基于tcga-crc数据集筛选高频变异基因,并经过“预后”and“结直肠癌”and“药物”等关键词组合的知识库(pharmgkb、nccn、csco、fda、nmpa、ema、ncbipubmed)过滤,得到581个候选标志物基因。
s1.3.2采用步骤2.2.3所述基于遗传算法的进化迭代方法,筛选到包括45个基因及其变异的标志物组合,具体基因集包括:kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1;pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e。
s1.3.3对以上dna层面生物标志物组合,在tcga-crc数据集进行检验,利用timeroc评估该组合的准确性,1年、3年和5年的生存期预测准确率(auc)分别达到0.87、0.83和0.80。采用风险评分的中位数将样本分为高、低两组做km分析,结果显示低评分组患者预后更好(p-value=4.52e-8)。同时,在cptac-2coloncancer(110例)、icgc的coad-us(402例)和coad-cn(321例)数据集上进行生存期预测,基因组合准确率auc达到0.81.
s1.4结直肠肿瘤综合状态评估基因集及评分系统开发
s1.4.1确定4-dysreg中7个基因和筛选到的45个结直肠癌重要变异基因,共计53个基因,作为结直肠肿瘤综合状态评估基因集。
s1.4.2综合纳入结直肠癌病人的年龄、性别、手术情况(有/无)、病理级别(i-iv)和微卫星不稳定(msi)等作为结直肠肿瘤综合状态评分系统的输入信息。
s1.4.3根据步骤3.3多元信息的充分整合构建综合评分系统中所述方法,采用随机森林与权重投票器的机器学习方案,以python语言开发了结直肠肿瘤综合状态评分系统的原型。
s1.4.4该系统可以输出结直肠癌病人的综合状态评分,该评分同其预后和治疗手段(化疗、靶向、免疫抑制剂)等效果有一定相关性,可以提供参考指导。
对该评分系统解析可以发现,所有输入特征均对生存预后有所贡献;但对治疗手段效果预测的权重有所不同,其中runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1的贡献集中在化疗方案效果预测,kras、nras、braf、her2、kit、pdgfra、sdha、sdhb、sdhc、sdhd、nf1的贡献侧重于靶向治疗方案效果预测,而pd1、pdl1、pdl2、ctla4、tigit、tim3、lag3、ifng、ccl2、gzma、prf1、cxcl8、cxcl9、cxcl10、tgfb1、sox10、serpinb9、cd8a、cd8b、gzma、gzmb、prf1、ccl5、cd27、cd274、cmklr1、cxcr6、nkg7、ido1、psmb10、stat1、stk11、hla-dqa1、hla-drb1、hla-e侧重于结直肠肿瘤免疫浸润与免疫细胞毒性状态评估,对免疫抑制剂治疗方案效果预测贡献较大;手术情况(有/无)、病理级别(i-iv)和微卫星不稳定(msi)等信息对于预后和治疗效果预测均有贡献。
s1.5结直肠肿瘤检测panel设计与评估系统构建
s1.5.1梳理上述53个基因的检测信息(如rna表达值,拷贝数cnv,基因变异包括snp、indel、fusion等),然后通过ncbiofficename或hgncapprovedofficialsymbol系统确定标准基因名。
s1.5.2参考步骤4基因检测panel设计方法,完成结直肠肿瘤检测panel的设计,并根据pcr或高通量测序平台做相应优化,比如runx3、gpr15、p2ry8、snai3、tlr7、atoh1、siglec1等7个基因组成的小panel即dysreg4,可以用pcr检测表达量;所有53个基因,可以利用panel设计捕获相关序列,利用高通量测序技术进行检测。捕获效率一般在30%~60%之间,全部基因目标区域覆盖度不低于95%,因此可认定探针设计合格。
s1.5.3根据panel检测数值和病例临床信息输入方式,对结直肠肿瘤综合状态评分系统进行优化,比如小panel即dysreg4,就可以仅保留该panel相关的功能模块,删掉45个变异基因的相关特征及预测功能模块。相应基因检测panel和该软件系统中对应功能模块,可以组合为一套配套联用流程,包括但不限于试剂盒/软件,检测装置/数据处理一体机,试剂盒/检测装置/数据在线分析平台等形式,用于结直肠肿瘤病人综合状态评估,并辅助临床决策包括但不限于辅助手术方案、化疗方案和靶向治疗方案选择、免疫疗法参考、预后状态评估等。
- 还没有人留言评论。精彩留言会获得点赞!