一种基于图卷积神经网络的蛋白质自相互作用预测方法与流程
本发明涉及机器学习和生物信息学领域,具体涉及一种新型的蛋白质自相互作用预测方法。
背景技术:
蛋白质是生物体的基本构件,几乎参与细胞内所有的生物过程。探索蛋白质的结构与功能,对于理解生命活动、疾病治疗以及新药研发具有重要意义。研究表明,蛋白质通常不单独执行功能,而是通过与其他蛋白质的相互作用来共同完成一个特定的功能。随着质谱分析、酵母双杂交、蛋白质芯片技术以及染色体免疫共沉淀等高通量生物技术的快速发展,使得蛋白质相互作用数据大量累积。如何从海量蛋白质相互作用数据中挖掘其中隐藏的生物学知识,揭示生命过程所涉及的反应通路、调控机制以及分子组成等问题正面临新的挑战。
在蛋白质相互作用中,蛋白质与自身发生相互作用(sip)占据重要位置。证据表明蛋白质自相互作用在包括酶激活、基因表达调控、信号转导和免疫应答等重要生物学过程中发挥关键作用。例如,pereira-leal等人通过对大规模生物蛋白质复合物起源和进化分析发现,许多蛋白质复合物的进化最初是由自身的相互作用建立起来的。ispolatov等人发现蛋白质相互作用网络中存在大量的自相互作用蛋白质,其在细胞系统中发挥重要作用。此外,自相互作用蛋白质能够在不增加基因组大小的情况下,通过自身的相互作用来调节蛋白质功能,从而扩展它们的功能多样性。
技术实现要素:
本发明提供一种新型的基于图卷积神经网络的蛋白质自相互作用预测的方法。
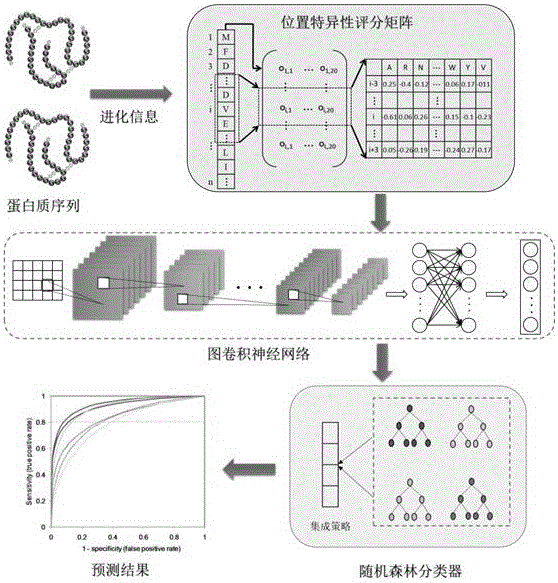
一种基于图卷积神经网络的蛋白质自相互作用预测的方法,包括以下步骤:
a、数据集的选择与建立:利用收集自uniprot、innatedb、biogrid、dip和matrixdb数据库的蛋白质序列数据构建预测蛋白质自相互作用的数据集;
b、位置特异性评分矩阵,pssm矩阵的构建;
c、图卷积神经网络的构建:利用深度学习的图卷积神经网络算法,将图卷积解释为概率度下嵌入函数的积分变换,通过逐层抽取的方式,将复杂的蛋白质进化信息数据转换为一系列简单的高级特征;
d、分类器模型的构建:利用随机森林算法,通过创建多个决策树的子分类器,并以集成的方式构建分类器模型。
其中,所述的步骤a,对得到的数据集需经过以下处理:
(1)从人类蛋白质组中删除长度大于5000或小于50个残基的蛋白质序列;
(2)被选择作为阳性样本的蛋白质数据至少满足以下条件之一:a.在uniprot中,蛋白质被定义为同寡聚体;b.至少有两篇已发表的文章报道过它们;c.至少通过两种大规模或一种小规模实验验证;
(3)阴性数据集中去除了所有已知的自相互作用蛋白质。
其中,所述的pssm矩阵为,将蛋白质序列转化为n*20的矩阵,其中n代表蛋白质的残基数,20代表为20个氨基酸种类,通过使用blast的位置特异性psiblast将每个蛋白质都转换成pssm矩阵。
其中,所述的步骤c为利用图卷积神经网络提取蛋白质自相互作用拓扑图的空间特征,其具体步骤为:
(1)从拓扑图中选择一个固定长度的节点序列;
(2)对序列中的每个节点,收集固定大小的邻域集合;
(3)对由当前节点及其对应的邻域构成的子图进行规范化,作为卷积结构的输入;
(4)通过逐层的卷积池化操作逐步提取出数据中隐藏的深度特征;
(5)利用梯度下降法最小化损失函数,对网络中的权重参数逐层反向调节,并通过多次迭代训练提高网络的精度。
其中,所述的步骤d,包括以下步骤:创建随机森林;基于创建的随机森林分类器进行预测;
所述的创建随机森林具体为:
(1)从全部m个特征中随机选择k个特征,其中k<<m;
(2)在k个特征中,用最佳分裂点计算节点d;
(3)用最佳分裂将节点分裂为子节点;
(4)重复前面三步的过程,直到获得i个数量的节点;
(5)重复第1到第4步n次创建n个树,从而形成一个森林;
所述的基于创建的随机森林分类器进行预测具体为:
(1)选取测试特征,用每个随机创建的决策树的规律去预测结果,并保存预测的结果;
(2)结算每个预测目标的得票数;
(3)将得票最多的预测目标作为随机森林算法的最终预测。
本发明的有益效果
本发明由得到的蛋白质位置特异性评分矩阵,即pssm矩阵,通过图卷积神经网络转化为具有代表性的简单的高级特征,最后通过随机森林进行预测。
其中,具体的,本发明基于卷积神经网络,利用深度学习的图卷积神经网络算法,将图卷积解释为概率度下嵌入函数的积分变化,通过逐层抽取的方式,将复杂的蛋白质进化信息数据转化为一系列简单的高级特征。
举例说明:
假设图g,的顶点集v,与概率空间(v,,f,p)相关联。对于图g,的子集g,其顶点是由概率度p生成的v,的i.i.d.样本,其函数的推广可描述如下:
此处,函数h(l)为来自l层的嵌入函数,u和v是独立随机变量。损失函数l是g(h(m))的期待,其可以表示为:
因此,可以使用t1的i.i.d.样本u1(i),…,ut1(i)来近似估计l层的积分变换,其表达式如下:
公式中h(0)t0为h(0)。由此损失l便可转换为:
由此可见,有效提取了蛋白质数据的深度特征。需要处理的数据量大大减少,
再使用随机森林分类器预测,极大的减少了计算数据的计算量,但是大大提高了预测进度。也克服了现有技术中,使用单一分类器(本发明为随机森林),导致构筑的模型预测精度差,推广能力差。
本发明计算代价小,功耗低;能有效预测潜在的蛋白质自相互作用,预测准确率可以达到90%以上。
附图说明
图1为本发明蛋白质自互相作用预测模型流程图;
图2为本发明在人类数据集上生成的五折交叉验证的roc曲线;横坐标0.1处,从上到下依次为:5thfold、4thfold、2thfold、1thfold、3thfold;
图3为本发明在酵母数据集上生成的五折交叉验证的roc曲线;横坐标0.2处,从上到下依次为:3thfold、4thfold、1thfold、2thfold、5thfold;
图4为不同分类器模型人类数据集上的比较;
图5为不同分类器模型酵母数据集上的比较。
具体实施方式
步骤a:本发明使用的人类和酵母数据集从uniprot、innatedb、biogrid、dip和matrixdb数据库中收集的能够与自身发生相互作用的蛋白质序列数据。
这些数据经过了如下的处理:(1)从人类蛋白质组中删除长度大于5000或小于50个残基的蛋白质序列;(2)被选择作为阳性样本的蛋白质数据至少满足以下条件之一:(a)在uniprot中,蛋白质被定义为同寡聚体;(b)至少有两篇已发表的文章报道过它们;(c)至少通过两种大规模或一种小规模实验验证;(3)阴性数据集中去除了所有已知的自相互作用蛋白质。
经过上述处理,我们筛选出了1441对自相互作用蛋白质和15938对非自相互作用蛋白质作为实验数据集。此外,为了进一步评估模型,我们使用同样的策略创建了酵母数据集,其包括710对自相互作用蛋白质和5511对非自相互作用蛋白质。
步骤b:本发明使用的位置特异性评分矩阵(pssm)是由gribskov等人提出的一种序列矩阵,其能根据序列或结构相似性预先排列的一组序列生成评分矩阵来检测远亲相关蛋白质。pssm将蛋白质序列转化为n*20的矩阵,可用公式表示如下:
此处,
在实验中,我们利用位置特异迭代blast(psi-blast)工具,通过比较swissprot数据库中的同源蛋白质,提取蛋白质序列的进化信息来生成pssm矩阵。为了得到同源序列,在实验中我们将psi-blast的参数e值和迭代次数分别设置为0.001和3。swissprot数据库和psi-blast工具可以从http://blast.ncbi.nlm.nih.gov/blast.cgi处下载。
步骤c:图卷积神经网络的构建:利用深度学习的图卷积神经网络算法,将图卷积解释为概率度下嵌入函数的积分变换,通过逐层抽取的方式,将复杂的蛋白质进化信息数据转换为一系列简单的高级特征,其维度由最初的400维原始数据抽取为精炼后的256维高级特征。
步骤d:使用随机森林算法,通过创建多个决策树的子分类器,并以集成的方式构建分类器模型。
为了说明本发明预测方法的效果,我们在基准数据集上对模型性能进行了评估。表1汇总了本发明在人类数据上生成的五折交叉验证结果。从表中可以看出,本发明的预测准确率为93.65%,特异性为99.64%,f1评分为37.11%,马修斯相关系数为43.01%,auc为60.68%。这些评价标准的标准偏差分别为0.64%、0.35%、10.54%、9.04%和4.96%。表2列出了本发明在酵母数据上生成的五折交叉验证结果。由表2可见,本发明的平均准确率为90.69%,特异性为99.08%,f1评分为38.37%,马修斯相关系数为41.19%,auc为64.30%。这些评价标准的标准偏差分别为0.50%、0.43%、4.63%、5.69%和2.97%。gcnsp模型在人类和酵母数据集上生成的五折交叉验证的roc曲线如图1和图2所示。
为了进一步评价分类器对模型整体性能的影响,我们将其与极限学习机(elm)和k近邻(knn)两个优秀的分类器模型进行比较。我们在实验中使用相同的特征提取方法,只替换模型中的分类器,并在人类和酵母数据集上进行了验证。
表3和表4分别汇总了elm和knn分类器模型在人类和酵母数据集上五折交叉验证的结果。从表3中可以看出,elm分类器模型在人类数据集上取得了87.19%的预测准确率,其五次实验的结果分别是86.88%、86.99%、88.26%、86.62%和87.21%;knn分类器模型取得了87.20%的预测准确率,其五次实验的结果分别是87.34%、87.63%、87.17%、86.30%和87.55%。而本发明取得了93.65%的预测准确率,分别比它们高了6.46%和6.45%。我们从表4中可以看到,elm和knn分类器模型在酵母数据集上分别取得了79.68%和82.86%的预测准确率,比本发明分别低了11.01%和7.83%。在其他评估参数中,本发明也取得了最优的实验结果。为了方便直观比较,我们将所有评估参数的结果以柱状图的形式进行展示,其结果如图3和图4所示。
近年来,一些优秀的预测蛋白质自相互作用的方法被提出,包括pspel、spar、slipper、ppievo和locfuse。为了更清楚地评估本发明的预测能力,我们将其与这些方法在人类和酵母数据集上进行了比较。表5和表6总结了上述方法在人类和酵母数据集上产生的实验结果。从表5可以看出,本发明在人类数据集的预测精度最高,比第二高spar方法高1.56%,比平均结果高5.84%。在评价标准特异性和mcc上,本发明也取得了最好的结果,分别比其他5种方法的平均值高出20.31%和12.76%。
从表6可以看出,本发明在酵母数据集的预测精度也达到了最佳,比其他五种方法的平均值高出16.96%,在评价标准特异性和mcc方面也取得了最好的结果,分别比其他5种方法的平均值高出18.27%和18.91%。对比结果表明,本发明具有良好的预测性能,比现有方法具有更高的预测精度。
最后应说明的是:以上所述仅为发明的优选实施例而已,并不用于限制发明,尽管参照前述实施例对发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!