一种无创产前检测装置的制作方法
1.本发明涉及生物信息学技术领域,具体地说,涉及一种无创产前检测装置。
背景技术:
2.无创产前检测(noninvasive prenatal testing,简称nipt)是一项非侵入性的孕期产检技术,这项技术是建立在孕妇外周血中存在着胎儿游离的dna这一科学发现的基础上的检测技术,具有很高的检测准确性,同时可以避免绒毛活检术、羊膜穿刺术和经腹静脉穿刺术等侵入性检测所带来的流产和宫内感染风险。
3.nipt检测技术原理是通过抽取孕妇外周血,提取出血浆游离dna,构建二代测序文库,利用二代测序仪得到孕妇血浆游离dna的序列信息。利用z检验方法比较染色体的含量与阴性对照集的差异,从而得到胎儿患病的风险。然而,测序试验中可以观察到批次效应,它的原因包括试验分析操作人员,时间、平台、实验室环境等。由于这些非生物因素的影响容易产生批次效应。如果不消除批次效应,对分析结果会产生很大的影响,容易造成假阴或者假阳。同时,位于灰区的样本也会增加,需对数据重新验证,从而增加检测成本及检测周期。cbs是目前检测cnv断点的主流算法之一。特点是分段结果稳定,对低浓度的断点检测灵敏性高。但是也有几个缺点:对波动较大的数据分段过于琐碎;没有提供是否属于cnv的信息;对于不同的数据需要调试不同的阈值。
4.因此,需要提供一种新的无创产前检测装置以解决现有技术的问题。
技术实现要素:
5.针对已有的nipt检测技术的以下缺点:
6.1、未考虑批次偏差的影响,造成假阴或者假阳;
7.2、若应用本批次样本作为对照参考集,样本数较少,导致染色体含量的sd偏大,则z值偏小,容易造成假阴;
8.3、灰区样本数较大,检测成本较高及检测周期较长。
9.本发明提出了一种有效降低批次偏差、检测周期短、检测精度高的无创产前检测装置。
10.为了实现本发明的发明目的,本发明的技术方案如下:
11.一种无创产前检测装置,所述装置包括:检测模块、数据质控及预处理模块、数据校正及处理模块和判断模块;
12.所述数据校正及处理模块:用于将待测孕妇样本基因组通过质控、窗口划分后获得的每个bin的ratio中的重复序列和群组cnv剔除后,进行gc校正和不同染色体基线带来的偏差校正,获得每个bin的含量x,并进一步根据动态数据库再次重新校正不同染色体基线带来的偏差;
13.所述动态数据库的构建方法如下:
14.根据整个待测样本批次中的t个待测样本s的每个bin的含量x对参考数据库中的
所有样本r进行筛选,选择参考数据库中d(s,r
j
)最小的男性样本a个和女性样本b个,组建针对本批次待测样本常染色体的动态数据库,其中男女性别比例无统计学差异,a+b不小于1000;选择参考数据库中d(s,r
j
)最小的男性样本a个和女性样本b个,分别组建针对本批次待测样本性染色体的动态数据库;d(s,r
j
)的计算公式如下:
[0015][0016]
其中,s代表所有待测样本,r
j
代表参考数据库中的第j个样本,代表t个待测样本中的第l个样本的第i条染色体bin的含量x的均值,代表参考数据库中第j个样本的第i条染色体的bin的含量x的均值。
[0017]
本发明优化了批次偏差校正的方法,通过特定的动态数据库构建方法获得了针对每一批次待测样本的动态数据库。利用本发明动态数据库有助于选择与批次波动类似的样本,较小批次偏差,提升阴性参考集的效果,使得检测准确性更高。其中,a+b不小于1000,可避免sd偏大,进行z统计检验时造成假阴的情况。
[0018]
为了避免一些重复序列的干扰,本发明在gc校正前对其进行了剔除(去除包含基因组串联重复及散在重复序列的bin)。另外,由于参考基因组hg19是欧洲人,在针对不同人群检测时,会存在一些种群水平上的假定cnv,本发明还对群组cnv进行了剔除。具体可通过叠加所有阴性数据库样本的窗口的reads数,以t检验法剔除离群点(群组cnv)。
[0019]
除了三体信号等其他干扰因素,gc含量也会对ur ratio造成较大的影响。gc校正可采用平滑样条法(统计每个窗口bin的reads数,并计算人类参考基因组中参考样本对应窗口的gc含量,以通过平滑样条法进行gc校正),尽可能减少误差干扰,有效地判断ur ratio的值是否有统计学意义上的异常。另外,不同染色体区域有不同的基线覆盖度,尽管gc校正减轻了基线差异,但是远不能消除基线之间的差异。因此,本发明还对不同染色体基线带来的偏差进行了校正。
[0020]
本发明中,所述数据校正及处理模块还用于在再次重新校正不同染色体基线带来的偏差后,进行pca降噪;
[0021]
所述pca降噪方式为:
[0022]
分别针对待测样本的常染色体和性染色体,以对应的动态数据库中的每一个阴性参考集样本的数据进行矩阵构建,具体以n个阴性参考集样本的m个bin构造一个n*m的矩阵x,其中每一项都是阴性参考集样本中bin的残差x,首先将x的每一行进行中心化,即减去每行的均值,计算中心化后矩阵x的协方差矩阵xx
t
,利用特征值分解矩阵得到特征值与特征向量w,将待测样本的残差x,构建1*e的矩阵t,旋转到相同的基上获得t',t'=tw,通过减去前k个主成分重建测试集x';e为待测样本的bin的个数,e=m;
[0023]
k值的确定:
[0024]
利用pca降噪后数据的残差与降噪前数据的残差的比值计算信息损失量l,评估降噪后数据信息丢失的情况来确定k值,具体步骤如下:
[0025]
将多例通过质控的已知核型结果的阳性样本基因组按100kb进行窗口划分后合并临近窗口获得窗宽为2mb的窗口,设置不同的k值,计算阳性区域的信息损失量l,损失量l的计算公式如下:
[0026][0027]
x
i
代表窗宽为2mb时的bin的ratio的均值,x
′
i
代表窗宽为2mb时进行所述pca降噪后bin的ratio的均值,n代表阳性区域为2mb窗口的个数;当信息损失量小于0.05时,所对应的k值最终用于待测样本的pca降噪。
[0028]
本发明中,所述数据校正及处理模块进行不同染色体基线带来的偏差校正方式为:
[0029]
通过计算相应数据库中全部正常对照样本的gc校正后的bin的含量的均值和标准差,构建加权线性回归模型,以对应位置校正后bin的ratio的标准差为权重,获得的残差x即为校正后的bin的含量x。
[0030]
本发明中,所述数据校正及处理模块进一步用于在进行所述pca降噪后,进行母源cnv识别并剔除不会遗传给后代的母源cnv;
[0031]
具体识别方式为:当多个连续的经pca降噪后的bin的含量x的绝对值abs(x)>0.5时,将这些bin合并确定为母源cnv位置,并计算所述母源cnv区域内所包含bin的含量的均值,即为所述母源cnv区域的杂合比hh;
[0032]
具体剔除方式为:当所述母源cnv区域的杂合比hh=1
‑
ff时,则将所述母源cnv区域予以剔除,当hh=1时,则将所述母源cnv区域予以保留;其中ff为待测样本的胎儿浓度。
[0033]
由于母体dna在血浆中的比例远大于胎儿的dna,因此本发明通过设置阈值来对母体cnv进行检测。
[0034]
本发明中,所述数据校正及处理模块还用于在剔除不会遗传给后代的母源cnv区域后,利用滑窗方法计算每个窗口的z值,并利用cbs算法对每个窗口的z值进行分割,检测cnv断点,再根据检测到的cnv断点获得分割后的cnv片段,依次计算所述分割后的cnv片段的杂合比和z值。本发明中,z值的计算公式如下:
[0035][0036]
其中,r
sample
代表待测样本每个窗口的ratio或分割后的cnv片段的ratio,每个窗口的ratio为该窗口包含的所有100kb的bin的含量的均值,每个分割后的cnv片段的ratio为该cnv片段包含的所有窗口的bin的含量的均值,meanr
reference
代表相应的动态数据库中阴性参考集样本与待测样本相对应的窗宽的ratio的均值,sdr
reference
代表相应的动态数据库中阴性参考集样本与待测样本相对应的窗宽的ratio的标准差。
[0037]
本发明中,每个窗口的窗宽为2mb,步长为50kb。
[0038]
本发明通过滑窗方法计算z值,从而增强了异常信号,并且通过增大窗口使得数据更加稳定,从而提升了检测结果准确性。
[0039]
本发明中,所述判断模块:用于根据所述数据校正及处理模块计算得到的分割后的cnv片段的杂合比和z值进行判断;当分割后的cnv片段的杂合比超出阈值[
‑
1/2*ff,1/2*ff],且该分割后的cnv片段的z值的绝对值|z|大于等于3时,判为异常,否则判定为无异常;当判为异常后,若该分割后的cnv片段的z值大于等于3,则判定为重复,z值小于等于
‑
3,则判定为缺失。
[0040]
本发明中,所述检测模块:用于对孕妇外周血游离dna进行高通量测序,获得待测孕妇样本基因组。
[0041]
本发明中,所述数据质控及预处理模块:用于将测序得到的待测孕妇样本基因组去除接头及低质量的reads,然后与人类基因组hg19进行比对,剔除未比对上、重复比对及比对到多个位置的reads,仅保留唯一比对上的reads,若unique reads数大于35m,总的gc含量在38%~42%之间,则通过质控;之后,进行窗口划分,窗宽100kb,重叠区域为50kb。
[0042]
由于nipt样本测序覆盖度很低,本发明采用基于划分窗口的方法,从而来减小数据的波动并有助于进行gg校正。
[0043]
本发明的有益效果至少在于:
[0044]
1.应用动态数据库,有效的降低了批次偏差造成的影响,避免了由于批次偏差导致的假阴或者假阳;还避免了应用待测批次样本做参考集,从而避免由于样本量少,造成的结果不准确;
[0045]
2.减少了灰区样本数量,降低了检测成本,缩短了检测周期;
[0046]
3.降低了cnv检测的假阳性,提高了检测精度。
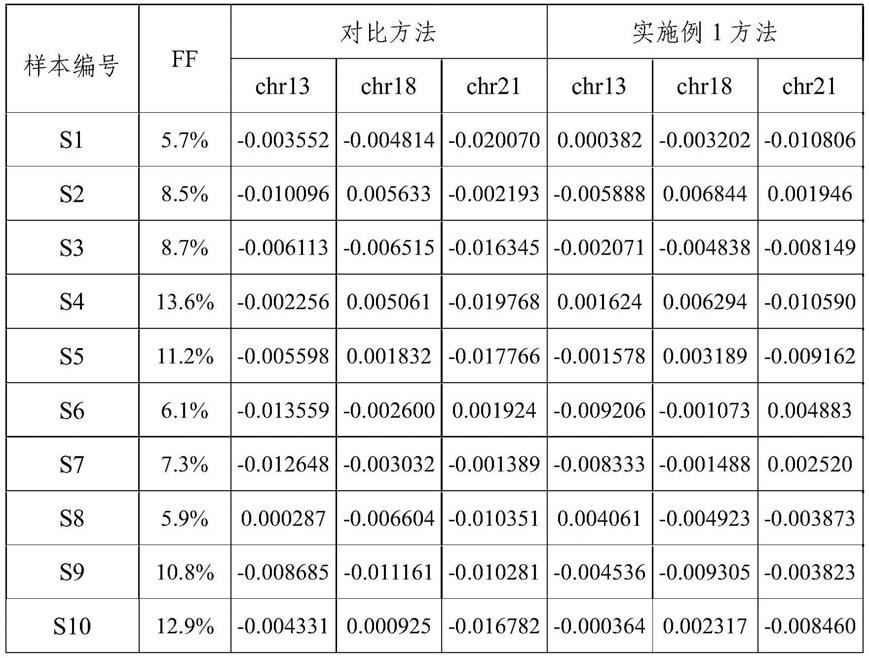
具体实施方式
[0047]
下面将结合实施例对本发明的优选实施方式进行详细说明。需要理解的是以下实施例的给出仅是为了起到说明的目的,并不是用于对本发明的范围进行限制。本领域的技术人员在不背离本发明的宗旨和精神的情况下,可以对本发明进行各种修改和替换。
[0048]
下述实施例中所使用的实验方法如无特殊说明,均为常规方法。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0049]
实施例1
[0050]
本实施例提供一种以本发明的无创产前检测装置进行染色体变异检测的方法。
[0051]
所述装置包括:检测模块、数据质控及预处理模块、数据校正及处理模块和判断模块。具体步骤如下:
[0052]
1、阴性参考集构建
[0053]
(1)样本选择
[0054]
选取9000例孕周大于等于12周且核型分析无染色体异常的孕妇样本,男和女比例无统计学差异。提取游离dna,按高通量方法进行基因组测序,采用单端测序方式获取读长50bp的fastq数据,所用测序平台为华大智造mgiseq
‑
2000基因测序仪。
[0055]
(2)数据预处理
[0056]
将测序数据去除带接头的reads,碱基质量小于5的碱基数大于总碱基数50%的reads,及n碱基数大于总碱基数5%的reads,保留剩余的reads。
[0057]
(3)比对定位序列位置及质控
[0058]
将过滤后的reads比对到人类基因组参考序列hg19上,剔除未比对上的reads,重复比对的reads,及比对到多个位置的reads,仅保留唯一比对上的reads,要求数据量大于35m。统计总的gc含量,要求gc含量在38%~42%之间。
[0059]
(4)分窗统计unique reads数
[0060]
将阴性参考集的每条染色体分窗统计,窗宽为100kb,滑动步长为50kb。
[0061]
去除包含基因组串联重复及散在重复序列的bin,通过叠加所有阴性数据库样本的窗口的reads数,t检验法剔除离群点,以去除群组cnv所对应的bin。
[0062]
(5)gc校正
[0063]
统计每个窗口bin的reads数,针对人类参考基因组hg19,根据窗宽的划分,计算每个窗口对应的gc含量。利用平滑样条法分别进行校正。之后根据9000例阴性对照样本,计算每个bin的均值和标准差,构建加权线性回归模型,权重为对应位置校正后bin的ratio的标准差,从而校正不同染色体基线带来的偏差,计算出每条染色体的残差x,即为阴性对照样本校正后bin的含量。
[0064]
(6)动态数据库构建
[0065]
根据整个待测样本批次中的t个待测样本s的每个bin的含量x对参考数据库(阴性数据库)中的所有样本r(9000例)进行筛选,根据d(s,r
j
)的计算公式,选择参考数据库中d(s,r
j
)最小的男性样本500例和女性样本500例,组建针对本批次待测样本常染色体的动态数据库;选择参考数据库中d(s,r
j
)最小的男性样本500例和女性样本500例,分别组建针对本批次待测样本两个性染色体的动态数据库(男、女各一个)。
[0066]
d(s,r
j
)的计算公式如下:
[0067][0068]
其中,s代表所有待测样本,r
j
代表参考数据库中的第j个样本,代表t个待测样本中的第l个样本的第i条染色体bin的含量x的均值,代表参考数据库中第j个样本的第i条染色体的bin的含量x的均值。
[0069]
其中,通过阈值来判断待测样本的性别,所述阈值由如下方法获得:统计大量流产组织样本的h,h=2c/(1+c),其中,c为样本x染色体bin的ratio的均值与y染色体bin的ratio的均值之比。根据k均值聚类方法,获得所述阈值。本实施例中所述阈值为0.025。
[0070]
将待测样本x染色体和y染色体bin的ratio的均值,代入男胎比例的计算公式h中,当h≥0.025时为男胎,否则为女胎。
[0071]
分别根据所获得的常染色体的动态数据库和性染色体的动态数据库中的所有样本gc校正后的bin的ratio,计算每个bin的均值和标准差,重新构建加权线性回归模型,权重为对应位置校正后bin的ratio的标准差,分别计算出每条染色体的残差x,即为阴性对照样本重新校正后bin的含量;
[0072]
(7)pca算法提取主成分
[0073]
以所获得的常染色体的动态数据库中的1000例阴性参考集样本的m个bin构造一个1000*m的矩阵x,其中每一项都是阴性参考集样本中bin的ratio,窗宽为100kb时,m为61927。首先将x的每一行进行中心化,即减去每行的均值,计算中心化后矩阵x的协方差矩阵xx
t
,利用特征值分解矩阵得到常染色体的特征值与特征向量w。
[0074]
分别以所获得的两个性染色体的动态数据库中的每一个阴性参考集样本以上述方法获得两个性染色体的特征值与特征向量w。
[0075]
确定测试时所采用的k值:
[0076]
利用pca降噪后数据的残差与降噪前数据的残差的比值计算信息损失量l,评估降
噪后数据信息丢失的情况来确定k值,具体步骤如下:
[0077]
将300例通过质控的已知核型结果的阳性样本基因组按100kb进行窗口划分后合并临近窗口获得窗宽为2mb的窗口,设置不同的k值,计算阳性区域的信息损失量l,损失量l的计算公式如下:
[0078][0079]
x
i
代表窗宽为2mb时的bin的ratio的均值,x
′
i
代表窗宽为2mb时进行所述pca降噪后bin的ratio的均值,n代表阳性区域为2mb窗口的个数;当信息损失量小于0.05时,所对应的k值最终用于待测样本的pca降噪。本实施例中最终k值确定为8。
[0080]
2、从上述构建的动态数据库所对应的待测样本批次中选择待测孕妇样本,进行染色体异常分析:
[0081]
(1)通过检测模块对待测孕妇样本,提取dna,进行全基因组测序,得到fastq数据。
[0082]
(2)通过数据质控及预处理模块对测序数据进行去除接头及低质量的reads、剔除非唯一比对上的reads、质控、窗口划分,具体方式与上述阴性参考集构建时的数据处理步骤(2)~(4)中所述相同。
[0083]
进一步,通过数据校正及处理模块将窗口划分后获得的每个bin的ratio中的重复序列和群组cnv剔除,进行gc校正(具体方式与上述阴性参考集构建时的数据处理步骤(4)~(5)中所述相同)和不同染色体基线带来的偏差校正。
[0084]
不同染色体基线带来的偏差校正方法如下:根据上述9000例阴性对照样本校正后的bin的ratio(参见阴性参考集构建时的数据处理步骤(5)),计算每个bin的均值和标准差,构建加权线性回归模型,权重为对应位置校正后bin的ratio的标准差,从而校正不同染色体基线带来的偏差,计算出每条染色体的残差x,即为待测样本校正后bin的含量。
[0085]
(3)按照阴性参考集构建时的数据处理步骤(6)判断待测样本的性别。
[0086]
通过数据校正及处理模块根据上述构建的动态数据库再次重新校正不同染色体基线带来的偏差。具体根据针对本批次筛选得到的常染色体的动态数据库,计算校正后的bin的ratio的均值和标准差,重新构建加权线性回归模型,权重为对应位置校正后bin的ratio的标准差,计算出每条常染色体的残差x,即为待测样本重新校正后的常染色体的每个bin的ratio。根据针对本批次筛选得到的性染色体的动态数据库进行上述同样操作,计算出待测样本性染色体的残差x,即为待测样本重新校正后的性染色体的每个bin的ratio。
[0087]
(4)数据校正及处理模块根据上述阴性参考集构建时的数据处理步骤(7)得到的常染色和性染色体的特征值和特征向量w,分别将待测样本的残差x,构建1*e的矩阵t,旋转到相同的基上获得t',t'=tw,通过减去前8个主成分重建测试集x',e为待测样本的bin的个数,e=m。
[0088]
(5)通过数据校正及处理模块利用发表于美国acmg会刊《genetics in medicine》(august 2019 22(2),doi:10.1038/s41436
‑
019
‑
0636
‑
5,minghao dang;hanli xu;jingbo zhang,etc.)的文章《inferring fetal fractions from read heterozygosity empowers the noninvasive prenatal screening》所述的方法计算胎儿浓度ff。
[0089]
(6)以数据校正及处理模块进行母源cnv识别并剔除不会遗传给后代的母源cnv;
[0090]
具体识别、剔除方式为:当多个连续的经pca降噪后的bin的含量x的绝对值abs(x)
>0.5时,将这些bin合并确定为母源cnv位置,并计算所述母源cnv区域内所包含bin的含量的均值,即为所述母源cnv区域的杂合比hh;当所述母源cnv区域的杂合比hh=1
‑
ff时,则将所述母源cnv区域予以剔除,当hh=1时,则将所述母源cnv区域予以保留;其中ff为上述步骤获得的待测样本的胎儿浓度。
[0091]
以数据校正及处理模块将待测样本的每条染色体的bin按2mb的窗宽进行合并,步长为50kb,重新划分的2mb窗口bin的ratio为该窗口包含的所有100kb的bin的ratio的均值。计算相应动态数据库中阴性参考集样本的所有2mb窗口bin的ratio,并计算每个2mb窗口bin的均值及标准差。利用z检验公式,计算每个bin的z值。
[0092]
(7)以数据校正及处理模块利用环状二元分割算法(cbs,circular binary segmentation)根据上述计算的每个2mb窗口bin的z值识别染色体的断点,并根据断点计算每个分割后的cnv片段segment所包含的2mb窗口bin的ratio的均值,即该分割后的cnv片段segment的ratio(该分割后的片段的杂合比);并根据断点,计算相应动态数据库中所有阴性参考样本对应片段区域的ratio的均值和标准差,重新计算各分割后的cnv片段segment区域的z值。
[0093]
z值的计算公式如下:
[0094][0095]
其中,r
sample
代表待测样本每个窗口的ratio或分割后的cnv片段的ratio,每个窗口的ratio为该窗口包含的所有100kb的bin的含量的均值,每个分割后的cnv片段的ratio为该cnv片段包含的所有2mb的窗口的bin的含量的均值,meanr
reference
代表相应的动态数据库中阴性参考集样本与待测样本相对应的窗宽的ratio的均值,sdr
reference
代表相应的动态数据库中阴性参考集样本与待测样本相对应的窗宽的ratio的标准差。
[0096]
当计算每个窗口的z值时,r
sample
代表待测样本每个窗口的ratio,当计算各分割后的cnv片段的z值时,r
sample
代表待测样本分割后的cnv片段的ratio。
[0097]
当计算的窗口或片段在常染色体上时,相应的动态数据库为上述构建的常染色体的动态数据库;当计算的窗口或片段在性染色体上时,相应的动态数据库为上述构建的性染色体的动态数据库。
[0098]
(8)通过判断模块对各分割后的cnv片段进行判断,设segment片段的ratio阈值[
‑
1/2*ff,1/2*ff],超出阈值范围,且|z|大于等于3时,判为染色体异常,否则判定为无染色体异常。当判为异常后,若该分割后的cnv片段的z值大于等于3,则判定为重复,z值小于等于
‑
3,则判定为缺失。其中,ff为待测样本的胎儿浓度。
[0099]
实施例2
[0100]
本实施例利用实施例1所示的方法和对比方法对待测样本进行测试。
[0101]
(1)选择20例孕妇的外周血进行检测,编号为s1
‑
s20,核型结果显示:1名怀有21号染色体三体胎儿,其余样本均为阴性。
[0102]
(2)根据本发明实施例1的方法和对比方法对这20例样本进行测试。对比方法与实施例1的方法相同,区别仅在于:不进行动态数据库的构建和使用,也不再重新校正不同染色体基线带来的偏差,采用全部阴性参考集。在对比方法中,当同一批次中|z|大于等于3的样本数大于本批次总样本数据的一半时,且同一条染色体的z值均偏大或者偏小时,认为存
在批次偏差,不能直接判定样本的结果,需重建库验证。
[0103]
表1为各待测样本以实施例1和对比方法进行检测时获得的chr13/chr18/chr21的染色体含量,即经上述检测步骤(7)cbs算法获得的分割后的cnv片段segment的ratio(r
sample
)和ff。表2为各待测样本以实施例1和对比方法进行检测时获得的meanr
reference
、sdr
reference
。表3为各待测样本以实施例1和对比方法进行检测时获得的分割后的cnv片段(chr13/chr18/chr21)的z值。如表3所示,s18在两种方法(实施例1和对比方法)检测下,结果一致,均为阳性。其余样本以实施例1的方法检测判定均为正常,与核型结果一致。而以对比方法进行检测判定结果出现了多处z值异常(参见表中加粗的数据),可能由批次偏差导致,无法对待测样本结果直接进行判断。由此可见,本发明采用动态数据库的方法可以校正样本偏差,减少假阳性或者灰区样本。
[0104]
表1
[0105]
[0106][0107]
表2
[0108][0109]
表3
[0110]
[0111][0112]
实施例3
[0113]
本实施例利用实施例1所示的方法对待测样本进行测试。
[0114]
(1)选择30例孕妇的外周血进行检测,编号为t1
‑
t30,核型结果显示:19例21号染色体三体综合征,3例18号染色体三体综合征,1例13号染色体三体综合征,7例微缺失微重复综合征。
[0115]
(2)利用实施例1对各样本进行检测。
[0116]
表4为23例非整倍体异常样本检测结果(参见表中加粗的数据),全部与核型结果一致。表5为7例微缺失微重复综合征样本检测结果,全部与核型结果一致,从中可知胎儿浓度为0.052,片段大小约2m的区域也可准确检出。
[0117]
表4非整倍体异常结果
[0118]
样本编号ffratio_chr13ratio_chr18ratio_chr21z值_chr13z值_chr18z值_chr21核型结果t114.7%
‑
0.0020.0070.158
‑
0.912.3537.26t21t28.7%
‑
0.003
‑
0.0040.090
‑
1.32
‑
1.4321.23t21t38.1%
‑
0.008
‑
0.0030.074
‑
2.18
‑
0.9817.42t21t48.2%
‑
0.002
‑
0.0050.076
‑
0.59
‑
1.7917.98t21t511.8%
‑
0.003
‑
0.0020.110
‑
1.26
‑
0.5225.98t21t66.3%
‑
0.001
‑
0.0040.057
‑
0.40
‑
1.2813.59t21t77.7%
‑
0.001
‑
0.0020.067
‑
0.21
‑
0.5015.80t21t89.8%0.001
‑
0.0050.0920.55
‑
1.5521.85t21t912.8%0.0000.0000.1190.12
‑
0.0128.12t21t107.9%0.0010.0000.0770.350.0518.09t21t119.9%
‑
0.012
‑
0.0040.089
‑
2.55
‑
1.4021.11t21t1214.1%0.002
‑
0.0040.1710.71
‑
1.3140.39t21t138.9%
‑
0.007
‑
0.0030.070
‑
2.56
‑
0.9516.66t21
t146.3%
‑
0.004
‑
0.0050.049
‑
1.62
‑
1.7111.56t21t1512.1%
‑
0.006
‑
0.0010.118
‑
2.21
‑
0.3027.97t21t167.3%
‑
0.0030.0000.080
‑
1.26
‑
0.0918.94t21t177.6%
‑
0.003
‑
0.0020.067
‑
0.97
‑
0.4915.87t21t189.1%0.0020.0030.1000.911.2323.67t21t1910.6%
‑
0.0010.0010.097
‑
0.430.4022.86t21t2014.8%
‑
0.0010.145
‑
0.006
‑
0.1949.74
‑
1.38t18t216.3%
‑
0.0040.0530.001
‑
1.5318.180.22t18t226.0%
‑
0.0060.047
‑
0.007
‑
2.4116.08
‑
1.59t18t239.0%0.091
‑
0.0050.00035.65
‑
1.68
‑
0.10t13
[0119]
表5微缺失微重复样本检测结果
[0120]
样本编号染色体起始位置终止位置长度ratioz值ff缺失/重复核型结果t24chr22186750002155000028750000.112525.5715.10%重复22q11重复综合征t25chr2218675000215500002875000
‑
0.04331
‑
3.345.80%缺失22q11缺失综合征(digeorge综合征)t26chr2218275000205000002225000
‑
0.07558
‑
4.649.00%缺失22q11缺失综合征(digeorge综合征)t27chr2219150000215500002400000
‑
0.09636
‑
4.8112.90%缺失22q11缺失综合征(digeorge综合征)t28chr2218600000215500001950000
‑
0.04264
‑
3.125.20%缺失22q11缺失综合征(digeorge综合征)t29chr15237000002872500050250000.0808763.5610.20%重复15q11
‑
q13重复综合征t30chr41000001422500014125000
‑
0.07789
‑
4.9511.30%缺失wolf
‑
hirschhom综合征
[0121]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1