难溶性药物纳米混悬剂及其制备方法
本发明涉及医药
技术领域:
,具体涉及难溶性药物纳米混悬剂及其制备方法。
背景技术:
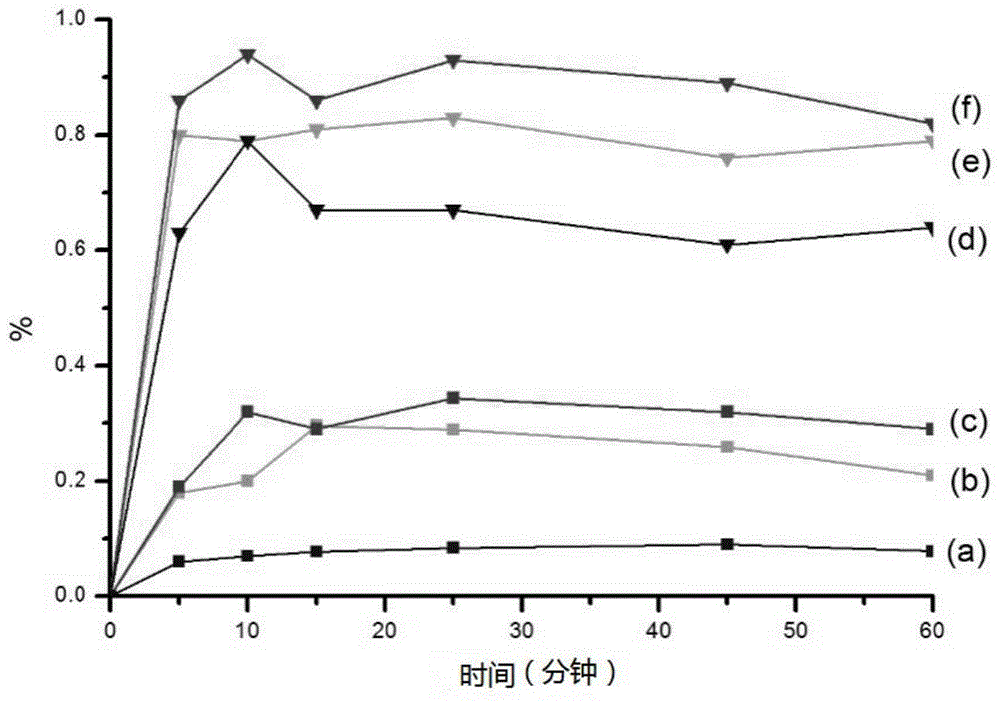
:近年来,随着组合化学、高通量筛选技术在药物研发中的广泛应用,涌现了很多分子量较大、结构复杂的化合物。根据统计,全球在售药物中40%是难溶性药物,而在研药物中高达90%。由于这些药物极低的水中溶解度,使其在胃肠道中溶出差,口服生物利用度低,限制了药物制剂开发和临床使用。纳米混悬剂是近年来针对难溶性药物开发的一种新型纳米给药系统,属于亚微米胶体分散体系,既可用于水难溶性药物,又适用于油、水均难溶性药物。相较于传统的混悬剂,纳米混悬剂的药物粒径小于1000nm,以纳米状态存在,能够增加药物溶出度,提高生物利用度,从而有利于人体吸收。此外,纳米混悬剂仅由药物和少量稳定剂组成,具有载药量高、用药剂量准确等优点,目前已成为药剂学领域研究的热点之一。溶剂-反溶剂法是目前制备纳米混悬剂的常用方法,具有可操作性强,制备工艺简单而被广泛应用。溶剂-反溶剂沉淀制备药物纳米混悬剂的原理依赖于药物在有机溶剂中具有较高的溶解度,再将含药有机溶剂加入反溶剂(通常是水或其他水性介质)中,形成过饱和水相,最后析出纳米级药物。然而,传统的反溶剂法制备纳米混悬剂采用的良溶剂大多为二氯甲烷、甲醇、dmso等有机溶剂,该类有机溶剂大多沸点较高,难以除去,制备成制剂后易造成有机溶剂的残留,且大多数有机溶剂有一定的毒副作用,长期使用对环境及人体不利。因此,寻找一种无毒、热稳定性好、可循环利用和价廉易得的绿色溶剂,代替传统的反溶剂法中的有机溶剂进行难溶性药物纳米混悬剂的制备具有重要意义。技术实现要素:本发明的目的是为了克服现有技术存在的现有溶剂-反溶剂法制备纳米混悬剂的过程中因有机溶剂残留导致对人体、环境产生不利影响的问题,提供一种难溶性药物纳米混悬剂及其制备方法,该方法提供一种无毒、热稳定性好、可循环利用和价廉易得的绿色溶剂代替传统的有机溶剂制备纳米混悬剂的方法,同时也是一种对环境友好型的纳米混悬剂的制备方法。为了实现上述目的,本发明一方面提供一种制备难溶性药物纳米混悬剂的方法,所述方法包括以下步骤:(1)将难溶性药物溶于潜溶剂中,得到药物-潜溶剂溶液;(2)将稳定剂溶于水中,得到含有稳定剂的水溶液;(3)在搅拌下,向步骤(2)得到的含有稳定剂的水溶液中加入步骤(1)得到的药物-潜溶剂溶液,加入完成后,继续搅拌和剪切得到难溶性药物纳米混悬剂;其中,潜溶剂与水的体积比为1:(2-10);所述稳定剂为为电荷稳定剂和立体稳定剂的混合物。优选地,所述难溶性药物为生物药剂学分类系统中ii类和ⅳ类药物中可溶于聚乙二醇的药物;优选地,所述难溶性药物选自芹菜素、布洛芬、吡仑帕奈、艾司利卡西平、盐酸特比萘芬、布地奈德、丙酸氟替卡松、丙酸倍氯米松和地塞米松中的至少一种。优选地,在步骤(1)中,所述潜溶剂为相对分子质量在200~600范围内的聚乙二醇。优选地,所述电荷稳定剂与水的重量比为0.1~0.3%;所述立体稳定剂与水的重量比为0.1~0.3%;进一步优选地,所述电荷稳定剂和所述立体稳定剂重量比为1:(1-3)。优选地,所述电荷稳定剂为十二烷基磺酸钠或十二烷基硫酸钠,所述立体稳定剂为泊洛沙姆或聚维酮k30。优选地,难溶性药物的重量与水的体积之比为0.0002~0.06g/ml。优选地,在步骤(3)中,药物-潜溶剂溶液的加入速度为20~80ml/min。优选地,在步骤(3)中,所述搅拌的速度为300~1100r/min。优选地,在步骤(3)中,所述剪切的时间为5~20min。本发明第二方面提供上述方法制备的难溶性药物纳米混悬剂,所述难溶性药物纳米混悬剂中药物的粒径为200~350nm。相比于现有难溶性药物纳米混悬剂的制备技术,本发明具有以下优点:1、本发明采用潜溶剂法进行纳米混悬剂的制备,不仅可以提高药物溶解度,进而提高载药量,而且潜溶剂自身能够作为稳定剂存在于纳米混悬体系中,使制备出的纳米混悬剂具有更高的物理稳定性。同时,本发明所用的聚乙二醇是一种无毒、热稳定性好、可循环利用和价廉易得的绿色溶剂,不仅能够消除传统反溶剂法对有机溶剂的依赖,避免使用有机溶剂带来的对人体、环境的不良影响。2、本发明联合使用电荷稳定剂(静电作用)和立体稳定剂(空间位阻作用),使得纳米混悬剂具有优异的稳定性。3、按本发明制备的纳米混悬剂,与原料药对比发现,其在ph1.0、4.5、6.8的缓冲液体外释放度,纳米混悬剂在各ph下60min时累积释放度达60%以上,而原料药在ph4.5下60min时累积释放度最高仅为29.08%。4、按本发明制备的纳米混悬剂,与原料药对比发现,其在大鼠体内cmax较原药增加3.8倍,auc0-t倍较原药增加1.6倍,绝对生物利用度较原药增加1.6倍,具有更快、更好的起效作用。5、在本发明中,无需冻干等固化操作,可以降低成本,有理由大生产操作。附图说明图1是测试例3中芹菜素和芹菜素纳米混悬剂溶出曲线对比图;图2是测试例4中两组灌胃给药的体内血药浓度随时间变化情况对比。具体实施方式以下结合附图对本发明的具体实施方式进行详细说明。应当理解的是,此处所描述的具体实施方式仅用于说明和解释本发明,并不用于限制本发明。在本文中所披露的范围的端点和任何值都不限于该精确的范围或值,这些范围或值应当理解为包含接近这些范围或值的值。对于数值范围来说,各个范围的端点值之间、各个范围的端点值和单独的点值之间,以及单独的点值之间可以彼此组合而得到一个或多个新的数值范围,这些数值范围应被视为在本文中具体公开。本发明一方面提供一种制备难溶性药物纳米混悬剂的方法,所述方法包括以下步骤:(1)将难溶性药物溶于潜溶剂中,得到药物-潜溶剂溶液;(2)将稳定剂溶于水中,得到含有稳定剂的水溶液;(3)在搅拌下,向步骤(2)得到的含有稳定剂的水溶液中加入步骤(1)得到的药物-潜溶剂溶液,加入完成后,继续搅拌和剪切得到难溶性药物纳米混悬剂;其中,潜溶剂与水的体积比为1:(2-10);所述稳定剂为为电荷稳定剂和立体稳定剂的混合物。在本发明中,所述难溶性药物可以为生物药剂学分类系统中ii类和ⅳ类药物中可溶于聚乙二醇的药物。在优选的实施方式中,所述难溶性药物选自芹菜素、布洛芬、吡仑帕奈、艾司利卡西平、盐酸特比萘芬、布地奈德、丙酸氟替卡松、丙酸倍氯米松和地塞米松中的至少一种。进一步优选地,所述难溶性药物选自芹菜素、布洛芬、吡仑帕奈、艾司利卡西平、盐酸特比萘芬、布地奈德、丙酸氟替卡松、丙酸倍氯米松或地塞米松。在优选的实施方式中,在步骤(1)中,所述潜溶剂为相对分子质量在200~600范围内的聚乙二醇。具体的,所述聚乙二醇的相对分子质量可以为200、300、400、500或600。在本发明中,使用潜溶剂法进行纳米混悬剂的制备。潜溶剂法是以溶剂-反溶剂法为基础,寻找一种对药物具有较高溶解度,稳定性好,绿色环保的助溶剂代替传统的有机溶剂制备纳米混悬剂的方法。使用潜溶剂不仅可以提高药物溶解度,进而提高载药量,而且潜溶剂自身能够作为稳定剂存在于纳米混悬体系中,使制备出纳米混悬剂具有更高的物理稳定性。同时,本发明所用的聚乙二醇是一种无毒、热稳定性好、可循环利用和价廉易得的绿色溶剂,还能能够消除传统反溶剂法对有机溶剂的依赖,避免使用有机溶剂带来的对人体、环境的不良影响。在优选的实施方式中,所述水为纯水。优选地,所述电荷稳定剂与水的重量比为0.1~0.3%;所述立体稳定剂与水的重量比为0.1~0.3%;进一步优选地,所述电荷稳定剂和所述立体稳定剂重量比为1:(1-3)。最优选为1:2。具体的,所述重量比可以为1:1、1:1.5、1:2、1:2.5或1:3。在本发明中,药物制成纳米混悬剂后其比表面积会增加,必然引起吉布自由能和表面能急剧增大,因此导致纳米混悬剂成为了热力学不稳定体系。为降低自身体系的能量,药物粒子即会自发地聚集,以减小其比表面积,从而出现一系列团聚、增长、沉降和晶型变化等不稳定物理现象。按照dlvo理论和空间稳定性理论,由于稳定剂的静电作用或空间位阻作用,可有效地吸附于药物粒子的表面,从而减少药物粒子团聚的发生。稳定剂的种类和用量将直接影响到纳米混悬剂的平均粒径、均匀性以及稳定性。因此,本发明通过使用联合稳定剂,即同时使用重量比为1:(1-3)的电荷稳定剂(静电作用)和立体稳定剂(空间位阻作用)时,纳米混悬剂的粒径在200~350nm之间,并且具有稳定性良好。在优选的实施方式中,所述电荷稳定剂为十二烷基磺酸钠或十二烷基硫酸钠,所述立体稳定剂为泊洛沙姆或聚维酮k30。在一种优选的实施方式中,所述稳定剂为十二烷基硫酸和钠聚维酮k30,并且十二烷基硫酸钠与水的重量比为0.1%,聚维酮k30与水的重量比为0.2%。在优选的实施方式中,所述潜溶剂与水的体积比为1:(2-10)。具体的,所述述潜溶剂与水的体积比可以为1:2、1:3、1:4、1:5、1:6、1:7、1:8、1:9或1:10。在本发明中,在采用聚乙二醇作为潜溶剂的同时,控制其与反溶剂(水)的体积比,使两者形成一定的体积差有利于药物析出而结晶,但当比例继续增大时,体系的载药量过低,不利于制剂药效发挥,故本发明中控制潜溶剂与反溶剂的比例在1:2~1:10之间。在优选的实施方式中,在步骤(3)中,药物-潜溶剂溶液的加入速度为20~80ml/min。具体的,所述加入速度可以为20ml/min、25ml/min、30ml/min、35ml/min、40ml/min、45ml/min、50ml/min、55ml/min、60ml/min、65ml/min、70ml/min、75ml/min或80ml/min。在优选的实施方式中,在步骤(3)中,所述搅拌的速度为300~1100r/min。具体的,所述搅拌的速度可以为r/min、400r/min、500r/min、600r/min、700r/min、800r/min、900r/min、1000r/min或1100r/min。在优选的实施方式中,在步骤(3)中,所述剪切的时间为5~20min。具体的,所述剪切的时间为5min、6min、7min、8min、9min、10min、11min、12min、13min、14min、15min、16min、17min、18min、19min或20min。在本优选的实施方式中,制备过程中难溶性药物的重量与水的体积之比为0.0002~0.06(g/ml)。在本发明中,在采用聚乙二醇作为潜溶剂的同时,控制其与反溶剂(水)的体积比为1:(2-10),使两者形成一定的体积差有利于药物析出而结晶,并且本发明采用电荷稳定剂和立体稳定剂两种协同作用,进一步防止药物聚集、增长等现象发生。本发明第二方面提供上述方法制备的难溶性药物纳米混悬剂,在本发明中,按照上述方法制得的难溶性药物纳米混悬剂中药物的粒径为200~350nm。以下将通过实施例对本发明进行详细描述,但本发明的保护范围并不局限于此。实施例1芹菜素纳米混悬剂的制备取1.6g的芹菜素原料药于20ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取160mg十二烷基硫酸钠(sds)和320mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于160ml纯水中充分溶胀。在持续搅拌的条件下,将芹菜素-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切15min得芹菜素纳米混悬剂。实施例2布洛芬纳米混悬剂的制备取4g的布洛芬原料药于20ml的聚乙二醇600(peg-600)中,沸水浴超声使其完全溶解;另取200mg十二烷基硫酸钠(sds)和400mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于200ml纯水中充分溶胀。在持续搅拌的条件下,将布洛芬-peg600溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为800r/min,待溶液滴加完全后,继续搅拌剪切10min得布洛芬纳米混悬剂。实施例3吡仑帕奈纳米混悬剂的制备取0.08g的吡仑帕奈原料药于20ml的聚乙二醇300(peg-300)中,沸水浴超声使其完全溶解;另取160mg十二烷基硫酸钠(sds)和320mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于160ml纯水中充分溶胀。在持续搅拌的条件下,将吡仑帕奈-peg300溶液通过蠕动泵加入含稳定剂的水中,注入速度为40ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切20min得吡仑帕奈纳米混悬剂。实施例4艾司利卡西平纳米混悬剂的制备取4.0g的艾司利卡西平原料药于20ml的聚乙二醇200(peg-200)中,沸水浴超声使其完全溶解;另取80mg十二烷基硫酸钠(sds)和160mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于80ml纯水中充分溶胀。在持续搅拌的条件下,将艾司利卡西平-peg200溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为1000r/min,待溶液滴加完全后,继续搅拌剪切10min得艾司利卡西平纳米混悬剂。实施例5盐酸特比萘芬纳米混悬剂的制备取1.2g的盐酸特比萘芬原料药于20ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取120mg十二烷基硫酸钠(sds)和120mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于120ml纯水中充分溶胀。在持续搅拌的条件下,将盐酸特比萘芬-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切5min得盐酸特比萘芬纳米混悬剂。实施例6布地奈德纳米混悬剂的制备取0.75g的布地奈德原料药于30ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取150mg十二烷基硫酸钠(sds)和300mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于150ml纯水中充分溶胀。在持续搅拌的条件下,将布地奈德-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切15min得布地奈德纳米混悬剂。实施例7丙酸氟替卡松纳米混悬剂的制备取0.0175g的丙酸氟替卡松原料药于35ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取70mg十二烷基硫酸钠(sds)和210mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于70ml纯水中充分溶胀。在持续搅拌的条件下,将丙酸氟替卡松-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为30ml/min,搅拌速度为600r/min,待溶液滴加完全后,继续搅拌剪切15min得丙酸氟替卡松纳米混悬剂。实施例8丙酸倍氯米松纳米混悬剂的制备取0.16g的丙酸倍氯米松原料药于40ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取200mg十二烷基硫酸钠(sds)和400mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于200ml纯水中充分溶胀。在持续搅拌的条件下,将丙酸倍氯米松-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为50ml/min,搅拌速度为750r/min,待溶液滴加完全后,继续搅拌剪切15min得丙酸倍氯米松纳米混悬剂。实施例9地塞米松纳米混悬剂的制备取1.4g的地塞米松原料药于35ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取280mg十二烷基硫酸钠(sds)和560mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于280ml纯水中充分溶胀。在持续搅拌的条件下,将地塞米松-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切20min得地塞米松纳米混悬剂。实施例10氯替泼诺纳米混悬剂的制备取1.4g的氯替泼诺原料药于35ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取280mg十二烷基硫酸钠(sds)和560mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于280ml纯水中充分溶胀。在持续搅拌的条件下,将氯替泼诺-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切15min得氯替泼诺纳米混悬剂。实施例11二氟泼尼酯纳米混悬剂的制备取0.1g的二氟泼尼酯原料药于25ml的聚乙二醇400(peg-400)中,沸水浴超声使其完全溶解;另取280mg十二烷基硫酸钠(sds)和560mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于200ml纯水中充分溶胀。在持续搅拌的条件下,将二氟泼尼酯-peg400溶液通过蠕动泵加入含稳定剂的水中,注入速度为20ml/min,搅拌速度为420r/min,待溶液滴加完全后,继续搅拌剪切15min得二氟泼尼酯纳米混悬剂。对比例1传统溶剂-反溶剂法制备纳米混悬剂取1.8g的芹菜素原料药于30ml的二甲基亚砜(dmso)中,沸水浴超声使其完全溶解;另取450mg聚乙烯吡咯烷酮k30(pvp-k30)作为稳定剂,溶解于240ml纯水中充分溶胀。在持续搅拌的条件下,将有机相通过蠕动泵加入水相,注入速度为20ml/min,搅拌速度为450r/min,待溶液滴加完全后,继续搅拌剪切15min得芹菜素纳米混悬剂。对比例2采用实施例2所述的方法进行制备,与之不同的是,聚乙二醇与水的体积比为1:11,也就是保持水的体积不变,将聚乙二醇醚的体积变为约18.2ml。对比例3采用实施例7所述的方法进行实施,与之不同的是,聚乙二醇与水的体积比为1:1,也就是说保持水的体积不变,将聚乙二醇的体积变为70ml。对比例4采用实施例1所述的方法进行实施,与之不同的是,不使用sds,pvp-k30的用量为480mg。对比例5采用实施例1所述的方法进行实施,与之不同的是,不使用pvp-k30,sds的用量为480mg。测试例1、采用激光粒度分析仪测定实施例和对比例制得的产品的平均粒径和pdi;仪器参数包括:通道宽度10μs,折光率1.333,激光波长632.8,散射角度90℃。检测结果如表1所示。表1编号实施例1实施例2实施例3实施例4实施例5实施例6粒径(nm)267.4±4.9215.2±2.8252.1±2.4312.4±1.8286.3±2.0301.5±1.5pdi0.0680.0760.0620.0750.0720.081编号实施例7实施例8实施例9实施例10实施例11粒径(nm)345.3±1.3291.6±1.7272.3±2.2280.3±1.9279.5±3.1pdi0.0890.0770.0790.0810.075编号对比例1对比例2对比例3对比例4对比例5粒径(nm)536.3±4.7195.4±1.7361.4±1.5536.3±4.7654.0±2.9pdi0.1560.0850.0910.3690.451由表1可知,按照本发明所述的方法制备的纳米潜溶剂,药物粒径在200~350nm范围内,比传统方法制备的纳米混悬剂的粒径小,药物粒度大小可以直接影响药物溶解度、溶解速度,进而影响到临床疗效,该粒径范围达到纳米级别,可显著提高药物溶解度和溶出速率,进而提高生物利用度,最终提升临床治疗效果。并且,随着潜溶剂和反溶剂的比例的增大,药物粒径呈减小的趋势。但当比例继续增大时,体系的载药量过低,不利于制剂药效发挥,因此控制潜溶剂和反溶剂的比例为1:(2-10)。2、对实施例和对比例制备的纳米混悬剂的稳定性进行考察,将上述样品置于25℃、湿度为60%的条件下,观察其含量、粒径和沉降体积比随时间的变化,检测结果如表2所示。表2由表2可知,按照本申请所述的方法进行制备,与传统制备方法相比较,不需要进行固化操作,得到的纳米混悬剂具有优异的稳定性。3、体外溶出度测定按照《中国药典》2020年版四部0931溶出度与释放度测定法第二法(桨法),对实施例1的产品和芹菜素原料进行溶出度测定。精密称取芹菜素原料5mg,纳米混悬剂0.2ml分别加入ph1.0、4.5、6.8的磷酸缓冲盐溶液1000ml,再加入0.5%吐温-80(5g)当溶出介质保持温度(37.0±0.5)℃下进行搅拌,搅拌桨转速设置为50r/min。分别于5、10、15、25、45、60min取样5ml(同时补加同体积的释放介质),用0.22μm微孔滤膜过滤,取续滤液3ml,甲醇定容到10ml,进液相色谱检测,连续3次,记录峰面积,外标法计算各时间芹菜素原药、纳米混悬剂的累计释放量。芹菜素和芹菜素纳米混悬剂溶出曲线对比如图1所示,其中,a,b,c为芹菜素原药在ph=1.0、4.5、6.8下的溶出曲线;d,e,f为芹菜素纳米混悬剂在ph=1.0、4.5、6.8下的溶出曲线。体外释放度考察结果如表3所示。表3由上述结果可知,本发明所制备实施例1的芹菜素纳米混悬剂在各ph下60min时累积释放度达60%以上,而芹菜素原料药在ph4.5下60min时累积释放度最高仅为29.08%,可见芹菜素纳米混悬剂的溶出速度和程度明显优于原料药。根据ostwald-freundlich方程和noyes-whitney方程可知,纳米混悬剂中药物的溶出度显著提高,芹菜素粒径减小,表面积增加,从而提高了累积释放度。4、体内药代动力学测定取实施例1样品和芹菜素原料药经大鼠口服给药后考察药物在体内的药动学特征,比较芹菜素纳米混悬剂和芹菜素原药的生物利用度。取雄性sd大鼠6只,随机分为3组:尾静脉组、原料药组以及实施例组,每组3只。尾静脉组采用尾静脉注射给药,给予芹菜素注射液,剂量为5mg/kg;原药组和纳米混悬剂组采用灌胃给药,分别给予芹菜素的口服混悬液和实施例1样品,剂量均为5mg/kg。试验前禁食12h,自由饮水,给药后2h统一进食。原药组和纳米混悬剂组灌胃给药,给药后0.25、0.5、1.0、2.0、3.0、4.0、6.0、8.0和24h采集血样;尾静脉组注射给药后于药后5min,0.25、0.5、0.75、1.0、1.5、2.0、4.0、8.0和24h采集血样;在以上设定时间点通过大鼠眼球后静脉丛眼眶取血0.30ml,置于含肝素抗凝管中,14000rpm离心5min,分离血浆,于-20℃冰箱中冷冻备用。采用高效液相色谱-质谱联用法测定芹菜素血药浓度,采用phoenix1.3软件(美国pharsight公司)的非房室模型计算给药后的药代动力学参数。绝对生物利用度两组灌胃给药的体内血药浓度随时间变化情况对比如图2所示,其中“powder-5mg/kg”为芹菜素原料药组,“nano-5mg/kg”为实施例组。体内药代动力学测试结果如表4所示。表4药动学结果表明,本发明所制备的芹菜素纳米混悬剂在大鼠体内的cmax较原药增加3.8倍,auc0-t较原药增加1.6倍,f(绝对生物利用度)较原药增加1.6倍。上述结果提示,本发明所制备的芹菜素纳米混悬剂较原药在药效的发挥上更具优势,具有更高的生物利用度。由上述测试结果可知,按照本发明所述的方法制备的纳米悬混剂,药物粒径在200~350nm范围内,具有优异的稳定性,并且悬混剂的溶出速度和程度相较于原料药明显提高,在大鼠体内具有更高的生物利用度。以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1