由生物质催化形成一氧化碳(CO)和氢(H2)的制作方法
本发明涉及通过下述制备一氧化碳(CO)和氢(H2)的方法:在浓酸的存在下,使来自任何源的生物质、生物质组分(例如木质素、木质纤维素、纤维素、半纤维素或其组合)或碳水化合物与多金属氧酸盐催化剂例如 H5PV2Mo10O40或其溶剂化物反应,以获得一氧化碳(CO);随后为氢(H2)的电化学释放。一氧化碳(CO)和氢(H2)可以任何所需比例组合,以获得合成气体 (合成气)。本发明还涉及由生物质、生物质组分和/或碳水化合物用于制备 H2、CO和甲酸/甲醛的方法。
背景技术:
环境关注和渐减量的化石燃料尤其是石油要求开发用于液体燃料的可再生资源,其对于运输和工业领域是关键的。太阳能和太阳能燃料已被吹捧为终极的无尽的能源,但日光的贮存及其以实际方式的用途仍是当今时代的重大挑战。
生物质具有作为可再生资源和基于太阳能的燃料的潜力,其中水和二氧化碳向葡萄糖并且随后向其他有机材料的转化,通过光合作用随后为进一步的生物合成途径来实现。因此,太阳能在陆生生物质中的贮存利用天然光合作用途径以及使用CO2和H2O作为碳和氢原子源的生物合成反应。陆生植物含有半纤维素~(C5H10O5)n和纤维素(C6H12O6)n,其可进行再加工以获得方便的烃燃料。通常,生物质含有35–50%纤维素、20–35%半纤维素和10–25%木质素。生物质的生物精炼是一个新兴领域,并且包括催化水解、溶剂分解、液化、热解、气化、氢解和氢化的过程都被加以考虑。(1,2)通过发酵将纤维素转化为D-葡萄糖(其唯一组分)且随后转化为乙醇以用作生物燃料可能是开发最充分的技术,然而,简单的酸无法选择性水解纤维素(3)仍是需要使用更昂贵的纤维素酶(cellusome)的问题。(4)水解/发酵方法还具有半纤维素及其主要水解产物D-木糖仍不能用于形成乙醇的缺点。另一方面,已提出纤维素/半纤维素水解作为到达化学品尤其是例如糖、甲酸、乙酰丙酸和羟甲基糠醛的途径。(5,6)已付出其他努力以将纤维素或纤维素衍生材料转化为烃 (7-9)、氢(10,11)和呋喃。(12)尽管有上述进展,反应选择性通常仍很低并且工艺方案是相当复杂的。
合成气体(也称为合成气)是主要由氢(H2)、一氧化碳(CO)和通常一些二氧化碳(CO2)组成的气体燃料混合物。合成气体是已知的工业商品,其用作中间构件块用于生产各种燃料,例如合成天然气、氨、甲醇和合成石油燃料。例如,烃可经由费歇尔-特罗普希法(Fischer-Tropsch process)由合成气体进行制备(13),或甲醇可作为燃料和燃料前体两者合成。(14)合成气体还可用作直接燃料源。在纯化状态下,合成气体的氢组分还可用于直接给氢燃料电池提供动力,用于发电和燃料电池电动车辆推进。氢还可通过哈伯-博施法(Haber-Bosch process)用于制备氨。
先前Khenkin等人已显示磷钒钼酸例如H5PV2Mo10O40多金属氧酸盐催化邻二醇和伯醇的碳-碳键切割。(15)在这类电子转移-氧转移反应中,来自多金属氧酸盐的氧原子被插入碳-碳键内,并且所释放的氢原子作为质子和电子保留在多金属氧酸盐上。例如,乙二醇氧化的初始产物是甲醛,并且1- 丁醇氧化最初获得甲醛和丙醛。Wasserscheid等人显示H5PV2Mo10O40可在相对高的O2压力(30巴)下,将在水中的碳水化合物例如半纤维素以50%得率转化为甲酸,伴随50%CO2的共同形成,但纤维素仅以低得率反应。(16,17) 氧化钒在该反应中是类似反应性的。(18)结果的分析显示甲酸的最大得率对于半纤维素为约50%,并且对于纤维素仅为约7%。(16-18)提出(16-18)甲酸还可转化为H2和CO2,通常在贵金属催化剂下,如先前描述的。(19,20)可替代地,酸催化的甲酸脱水为CO和H2O可加以考虑。(21)使用D-葡萄糖作为模型,这转化为潜在得率,在观察到的每摩尔D-葡萄糖的甲酸、3mol H2或CO但不是两者的50%得率。这远不是每摩尔D-葡萄糖的6摩尔CO和 H2两者的最佳值。
美国专利申请号US 2013/0245319公开了通过下述用于催化产生甲酸的方法:使α-羟基醛、α-羟基羧酸、碳水化合物或糖苷与通式[PMoxVyO40]q-的多金属氧酸盐离子催化剂反应,使用水作为溶剂。
Weinstock,I.A.等人(26)描述了使用Keggin型多金属氧酸盐(POM)和氧以漂白木浆用于制造纸。
美国专利号5,302,248、5,549,789和5,695,606描述了用于使木浆脱木素 /漂白用于制造纸的方法。漂白工艺涉及使木浆暴露于式 [Vl-MomWnNboTap(TM)qXrOs]x-的多金属氧酸盐,以产生水溶性氧化木质素,其随后通过将溶液加热至高温而氧化降解为CO2。
合成气体是用于形成在其他工业应用中的燃料和燃料前体的重要商品。仍存在未满足的对用于由生物质制备合成气体的有效方法的需要。
技术实现要素:
本发明涉及通过下述制备一氧化碳(CO)和氢(H2)的方法:在足以获得一氧化碳(CO)的条件下,在浓酸的存在下,使来自任何源的生物质、生物质组分(例如木质素、木质纤维素、纤维素、半纤维素或其组合)或碳水化合物与多金属氧酸盐催化剂反应;随后为氢(H2)的电化学释放。一氧化碳(CO)和氢(H2)可以任何所需比例组合,以获得合成气体(合成气)。本发明还涉及通过下述用于制备CO的方法:在足以获得一氧化碳(CO)的条件下,在浓酸的存在下,使生物质、生物质组分和/或碳水化合物与多金属氧酸盐催化剂反应。二氧化碳(CO2)可任选在这个过程中形成。本发明还涉及通过水煤气变换反应用于制备氢(H2)的方法,所述水煤气变换反应涉及在足以获得二氧化碳 (CO2)的条件下,在浓酸的存在下,使来自任何源的生物质、生物质组分或碳水化合物与多金属氧酸盐催化剂反应;随后为氢(H2)的电化学释放。本发明还涉及通过下述用于制备甲酸的方法:在足以获得甲酸的条件下,在选自醇或醇和水的混合物的溶剂中,使碳水化合物与多金属氧酸盐催化剂接触。多金属氧酸盐催化剂可为例如H5PV2Mo10O40或者其溶剂化物或水合物,或者本文描述的任何另外的多金属氧酸盐催化剂。
陆生植物含有70%半纤维素和纤维素,其是显著的可再生生物资源,具有作为碳基燃料的石油原料的替代物的潜力。碳水化合物的有效和选择性解构为其基础组分一氧化碳和氢(所谓的合成气体)是朝向这种潜力实现的重要关键步骤,因为由合成气体形成液体烃燃料是已知技术。本发明首次证实通过使用多金属氧酸盐作为电子转移-氧转移催化剂,通过最初形成的甲酸的脱水而切割所有碳-碳键来形成一氧化碳。在这种氧化还原反应中,氢原子作为质子和电子贮存于多金属氧酸盐上,并且可从多金属氧酸盐中作为氢电化学释放。共同形成合成气体。在氢经济情况下,该方法还可用于将一氧化碳转化为氢。此外,该方法并不限于碳水化合物,还可应用于其他生物质组分例如木质素和木质纤维素,以及作为整体或其任何部分的生物质。
有利地,在上述过程中,多金属氧酸盐溶液可再使用,而无需任何催化剂回收操作。因此,需要时,反应可重复至少一个另外的循环(例如10个或更多个循环),而无需再循环或回收溶剂。然而,需要时,可对多金属氧酸盐实施如本领域已知的回收/再循环方法。
本发明基于下述出乎意料的发现:在温和条件下,多金属氧酸盐催化剂例如H5PV2Mo10O40以高选择性和效率催化来自任何源的生物质或生物质组分或碳水化合物(包括但不限于生物质衍生的木质素、纤维素和半纤维素多糖(CnH2nOn)m,其中对于D-葡萄糖n=6,并且对于D-木糖n=5)的有效两步单罐转化为一氧化碳(CO)和氢(H2)。有利地,CO和H2可以任何所需比例组合,以获得合成气体(合成气)。如本文证实的,使用在作为溶剂的硫酸中的含钒多金属氧酸盐H5PV2Mo10O40作为催化剂,来自各种源的生物质,以及其个别组分木质素、半纤维素和纤维素被氧化为一氧化碳(CO),其中氢原子作为质子和电子贮存于多金属氧酸盐上。氢气(H2)从多金属氧酸盐中电化学释放,所述多金属氧酸盐返回到其原始氧化状态。以这种方式,生物质、木质素、纤维素和半纤维素定量转化为合成气体CO和H2,其可通过已知技术反应以获得烃。在另一个实施例中,CO经由一类水煤气变换反应原位氧化,以获得CO2和H2。
如本文考虑的,本发明人已发现在多金属氧酸盐催化剂的存在下,使生物质或生物质组分与浓酸反应导致一氧化碳(CO)形成,在一些情况下,通过甲酸(HC(=O)OH)和甲醛(HC(=O)H)的中间产物形成。甲酸/甲醛转化为CO 需要浓酸的存在下,即酸的浓度优选为在水中约80w/w%或更高。在一个优选实施例中,反应在纯酸(neat acid)中执行,其中酸用作反应的溶剂。氢(H2) 还可电化学生成。组合每个步骤的产物(即CO和H2)生成合成气作为最终产物。两步工艺(形成CO随后为H2)可有利地在单罐中同时或序贯执行。
对于合成气、甲酸或CO的产生,反应可在不存在氧(无氧)的条件下或在存在氧(有氧)的条件下进行。每种可能性代表本发明的一个分开实施例。
因此,在一个实施例中,本发明提供了用于由生物质或其组分或者碳水化合物制备一氧化碳(CO)和氢(H2)的方法,该方法包括下述步骤:(a)在足以产生一氧化碳(CO)和任选的二氧化碳(CO2)的条件下,在浓酸的存在下,使生物质、生物质组分或碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2)。反应可在有氧或无氧条件下进行。步骤(a)和(b) 可在单罐中同时执行,或可替代地,这些步骤可序贯执行,其中步骤(b)在步骤(a)之后。每种可能性代表本发明的一个分开实施例。
在一些实施例中,该方法还包括组合步骤(a)中产生的一氧化碳(CO)或其一部分和步骤(b)中产生的氢(H2)或其一部分,以便形成合成气体(合成气)。
通过净化反应混合物快速去除形成的CO可阻止其进一步氧化为CO2。因此,在一些实施例中,该方法还包括在步骤(a)后例如通过净化从反应中去除CO的步骤。
在另一个实施例中,本发明涉及用于由生物质或其组分或者碳水化合物制备一氧化碳(CO)和任选的二氧化碳(CO2)的方法,该方法包括下述步骤:在足以产生一氧化碳(CO)和任选的二氧化碳(CO2)的条件下,在浓酸的存在下,使生物质、生物质组分或碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触。反应可在有氧或无氧条件下进行。每种可能性代表本发明的一个分开实施例。
在一些实施例中,本发明的方法包括完整生物质的使用,所述完整生物质主要包含木质素、木质纤维素、纤维素和半纤维素。在其他实施例中,本发明的方法包括使用选自木质素、木质纤维素、纤维素、半纤维素及其任何组合的生物质组分。每种可能性代表本发明的一个分开实施例。
在一些实施例中,生物质可在执行本发明的方法之前分离成其组分中的一种或多种。在一些实施例中,生物质分离成半纤维素和木质纤维素,并且木质纤维素用于本发明的方法中。可替代地,生物质可分离成半纤维素和木质纤维素,木质纤维素还分离成木质素和纤维素,并且木质素或纤维素用于本发明的方法中。每种可能性代表本发明的一个分开实施例。
在其他实施例中,本发明的方法包括碳水化合物的使用。任何碳水化合物源均可用于本发明的工艺。非限制性例子包括单糖、二糖、三糖、寡糖、多糖、糖醇、植物生物质、淀粉及其任何组合。在一些实施例中,碳水化合物来源于陆生生物质,即纤维素、半纤维素或其组合。
酸优选以大于约50%、优选大于约80%、更优选大于约90%、更优选纯酸(约98%)的浓度使用,用作反应的溶剂。用于本发明的工艺中的合适酸包括但不限于H2SO4、HClO4、H3PO4、CH3SO3H、CF3SO3H、CF3COOH、 CH3COOH/H2SO4及其任何组合。每种可能性代表本发明的一个分开实施例。使用浓H2SO4即在水中约85%w/w或更大,例如在水中大约98w/w%H2SO4是目前优选的。
在一些实施例中,使用选自Au、Ag、Ni、Co、Pt、Pd、Cu、Al、Au/Ag、 Au/Cu、Au/Ag/Cu、Au/Pt、Au/Pd、Au/Ag/Cu/Pd、Pt-Rh、Ni-Co和Pt-Ni-Fe 电极的电极,进行工艺的步骤(b),即H2的电化学生成。Pt片电极的使用是目前优选的。
在另一个方面,本发明基于用于由碳水化合物制备甲酸的改良工艺的发现。生物质转化为甲酸先前已由其他人进行描述(US 2013/0245319,(16)),使用水作为反应溶剂。目前已预期地发现使用醇作为反应溶剂,含或不含水,显著增加反应的得率。因此,在另一个实施例中,本发明涉及用于由碳水化合物制备甲酸的方法,该方法包括下述步骤:在足以产生甲酸的条件下,在选自醇或醇和水的混合物的溶剂中,使碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触。醇优选甲醇,但也可为C2-C4醇。本文描述的碳水化合物中的任一种均可用于该反应中。反应可在有氧或无氧条件下进行。每种可能性代表本发明的一个分开实施例。
在另一个方面,本发明的方法可用于经由水煤气变换反应产生氢(H2)。已发现在生物质的转化期间CO部分氧化为CO2。因此,在一些实施例中,可进行工艺以实现CO至CO2的完全转化,随后为如上所述的氢的电化学释放,以便完成水煤气变换反应。因此,在另一个方面,本发明涉及用于由生物质或其组分或者碳水化合物制备氢(H2)的方法,该方法包括下述步骤:(a) 在足以产生二氧化碳(CO2)的条件下,在浓酸的存在下,使生物质、生物质组分或碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2);其中步骤(a)和(b)同时或序贯执行。上文描述的反应条件中的任一种包括生物质类型、酸等可用于该工艺。每种可能性代表本发明的一个分开实施例。
适用于本发明的多金属氧酸盐催化剂通常是由通式[XxMmOy]q-代表的多氧阴离子(polyoxoanion)盐或其溶剂化物,其中X是金属或非金属杂原子或者质子;M是选自钨(W)、钼(Mo)、铌(Nb)、钒(V)、钽(Ta)、铋(Bi)、锑(Sb)、锡(Sn)及其任何组合的增补原子(addenda atom);O是氧;x是0至6的整数; m是4至200的整数;y是5至1000的整数;并且q是0至30的整数。此类催化剂的非限制性例子在下文提供。在一个目前优选的实施例中,多金属氧酸盐催化剂是H5PV2Mo10O40。另外,多金属氧酸盐催化剂通常以溶剂化形式例如水合物。因此,本发明涵盖多金属氧酸盐溶剂化物,例如但不限于多金属氧酸盐水合物的使用。其他合适的多金属氧酸盐溶剂化物在下文详述中描述。
附图说明
本发明由与附图结合的下述详述将得到更全面理解和了解:
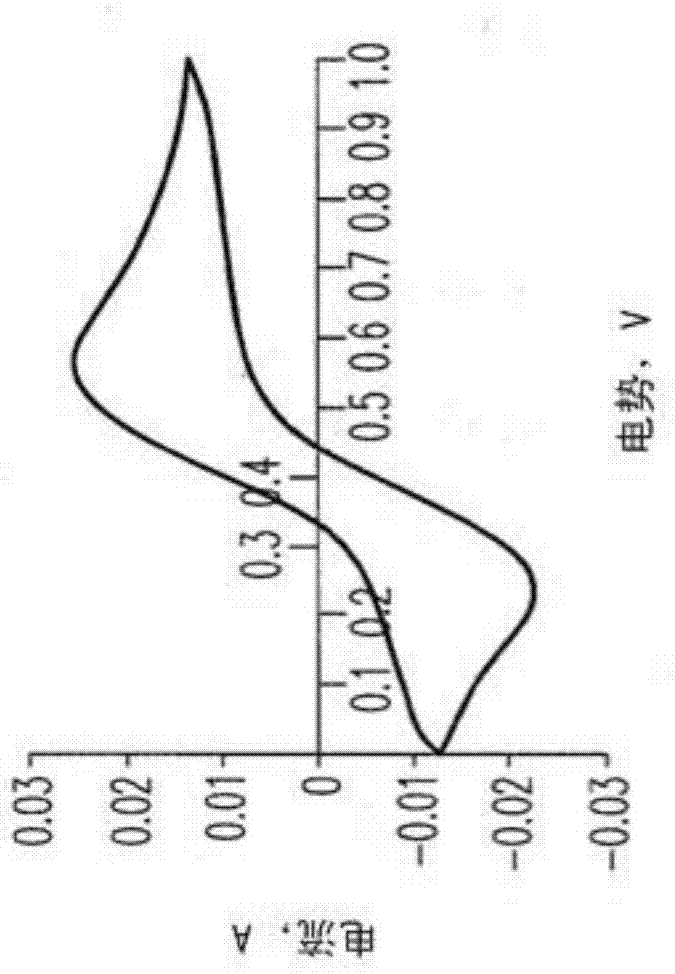
图1.反应溶液(在H2SO4中的0.42M H5PV2Mo10O40)的循环伏安图。 Pt用作参考、工作和对电极,扫描速率-200mV/秒。图1A-在纤维素氧化结束时,其为2.7个电子/H5PV2Mo10O40。图1B-在0.6V下氧化和多金属氧酸盐的25-30%氧化后,留下2.0个电子/H5PV2Mo10O40。
图2.纤维素的转化。纤维素至CO和H2的转化(图2A)以及CO的水煤气变换(图2B)。
具体实施方式
本发明充当由来自任何源的生物质或其组分或者碳水化合物形成一氧化碳(CO)和氢(H2)的新技术基础。分开形成的两种气体可以任何所需比例组合,以获得合成气体(合成气)。本发明还涉及用于由来自任何源的生物质或其组分或者碳水化合物制备一氧化碳(CO)的方法。本发明还涉及经由水煤气变换反应,用于由来自任何源的生物质或其组分或者碳水化合物制备氢 (H2)的方法。本发明还涉及用于由在选自醇或醇和水的混合物的溶剂中的碳水化合物制备甲酸的方法。
这些概念的所谓优点在于:(1)因为太阳能及其他能源(风能、地热能和核能)与其作为化学燃料的贮存相比较更简单地转化为电能,所以本文呈现的低温电化学方法具有相当大的总节能潜力。(2)合成气体得率可显著高于用生物质的高温蒸汽重整获得的那种,在生物质的高温蒸汽重整中使用来自空气的O2的需要导致CO2作为主要产物形成,其不由H2形成得到补偿,并且需要基于贵金属的催化剂用于定量转化。(3)本文呈现的概念指示需要1摩尔当量的多金属氧酸盐/碳原子;例如,在单罐反应中的10mg纤维素/780mg 多金属氧酸盐。然而,因为底物转化是定量的,所有产物均为气态的,并且催化剂在反应条件下是非常稳定的,所以本文方法可应用于涉及连续反应模式的工业用途,其中底物可连续供应并且所得到的气体(CO和/或H2)可使用固定量的催化剂连续去除。
定义
术语“合成气体”或“合成气”是主要由氢(H2)、一氧化碳(CO)和任选的一些二氧化碳(CO2)组成的混合物。
术语“浓酸”指以约50%或更大、优选大于约80%、更优选大于约85%、更优选大于约90%、最优选大于约95%的浓度存在的酸。酸充当反应的溶剂。本文描述的所有百分比指在水中的重量/重量(w/w)%。
生物质碳水化合物至甲酸、甲醛、CO、H2和合成气体的转化
在一个实施例中,本发明提供了用于由生物质制备一氧化碳(CO)和氢 (H2)的方法,该方法包括下述步骤:(a)在足以产生一氧化碳(CO)和任选的二氧化碳(CO2)的条件下,在浓酸的存在下,使生物质与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2)。
在另一个实施例中,本发明提供了用于由选自下述的生物质组分制备一氧化碳(CO)和氢(H2)的方法:木质素、木质纤维素、纤维素、半纤维素及其任何组合,该方法包括下述步骤:(a)在足以产生一氧化碳(CO)和任选的二氧化碳(CO2)的条件下,在浓酸的存在下,使生物质组分或组分组合与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2)。在一个实施例中,生物质组分是纤维素。在另一个实施例中,生物质组分是半纤维素。在另一个实施例中,生物质组分是木质素。在另一个实施例中,生物质组分是木质纤维素。在另一个实施例中,生物质组分是纤维素、半纤维素、木质素和木质纤维素的任何组合。在一些实施例中,生物质组分可从完整生物质中分离,或它可得自其他源例如合成或商业源。
在另一个实施例中,本发明提供了用于由碳水化合物制备一氧化碳(CO) 和氢(H2)的方法,该方法包括下述步骤:(a)在足以产生一氧化碳(CO)和任选的二氧化碳(CO2)的条件下,在浓酸的存在下,使碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2)。
在上述实施例的每一个中,一氧化碳(CO)和氢(H2)可以任何所需比例组合,以获得合成气体(合成气)。
在上述实施例的每一个中,步骤(a)和(b)可在单罐中同时执行,或可替代地,这些步骤可序贯执行,其中步骤(b)在步骤(a)之后。反应可在有氧或无氧条件下进行。每种可能性代表本发明的一个分开实施例。
本发明还可用于通过执行请求保护的工艺的步骤(a)且分离一氧化碳 (CO),不依赖于氢(H2)的形成而形成一氧化碳(CO)。因此,在一个实施例中,本发明涉及用于由生物质制备一氧化碳(CO)的方法,该方法包括下述步骤:在足以产生一氧化碳(CO)的条件下,在浓酸的存在下,使生物质与多金属氧酸盐催化剂或其溶剂化物接触。
在另一个实施例中,本发明涉及用于由选自下述的生物质组分制备一氧化碳(CO)的方法:木质素、木质纤维素、纤维素、半纤维素及其任何组合,该方法包括下述步骤:在足以产生一氧化碳(CO)的条件下,在浓酸的存在下,使生物质组分或组分组合与多金属氧酸盐催化剂或其溶剂化物接触。
在另一个实施例中,本发明涉及用于由碳水化合物制备一氧化碳(CO) 的方法,该方法包括下述步骤:在足以产生一氧化碳(CO)的条件下,在浓酸的存在下,使碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触。
在上述实施例的每一个中,反应可在有氧或无氧条件下进行。每种可能性代表本发明的一个分开实施例。
在另外一个实施例中,本发明涉及用于由碳水化合物制备甲酸的方法,该方法包括下述步骤:在足以产生甲酸的条件下,在选自醇或醇和水的混合物的溶剂中,使碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触。
多金属氧酸盐与生物质的比率可从约0.01到10当量多金属氧酸盐/底物中的碳原子不等,取决于催化剂和底物的性质。对于CO、合成气或甲酸的生产,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率优选为约1 当量或更少的多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。在一些实施例中,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率为约0.1-1当量多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。在其他实施例中,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率为约0.1当量多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。在其他实施例中,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率为约0.5当量多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。上述条件优选用于依照本发明的一些实施例的CO、合成气或甲酸形成。
不希望受任何特定理论和作用机制束缚,在碳水化合物的情况下,考虑如果醇用作反应溶剂,则本发明的工艺通过最初将碳水化合物转化为甲醛和甲酸以及相关的半缩醛/缩醛来实现。酸的存在还导致CO的形成,并且最终 H2通过使用例如Pt电极电化学生成。该工艺在下文方案1中就葡萄糖而言例示,但可应用于任何简单或复杂的碳水化合物,包括来源于陆生生物质的纤维素/半纤维素。
方案1
使用纤维素作为代表性底物的纤维素和半纤维素转化的表示在图2A中呈现,其中碳水化合物的电子转移-氧转移氧化连同最初贮存于多金属氧酸盐上的电子和质子的电化学氧化一起显示。
任何浓酸均可用于本发明的工艺中。非限制性酸包括H2SO4、HClO4、 H3PO4、CH3SO3H、CF3SO3H、CF3COOH、CH3COOH/H2SO4及其任何组合。每种可能性代表本发明的一个分开实施例。目前优选的酸是H2SO4。
在一些实施例中,使用本领域已知的电极,例如选自Au、Ag、Ni、Co、 Pt、Pd、Cu、Al、Au/Ag、Au/Cu、Au/Ag/Cu、Au/Pt、Au/Pd、Au/Ag/Cu/Pd、 Pt-Rh、Ni-Co和Pt-Ni-Fe电极的一种,通过多金属氧酸盐的电化学氧化来进行工艺的步骤(b),即H2的电化学生成。目前使用的电极是铂片电极。
水煤气变换反应
水煤气变换反应(27-29)从十八世纪就已知,但没有任何工业化,直到二十世纪初,当时人们认识到其用于产生氢以满足哈伯-博施法的需要的效用。水蒸气和一氧化碳之间的放热和可逆反应以产生氢和一氧化碳(CO+H2O →CO2+H2;ΔH=-41.1kJ/摩尔)被称为水煤气变换(WGS)反应。工业上, WGS反应在两个不同温度下进行:(a)使用基于Fe的催化剂(例如Fe2O3/Cr2 O3)的高温WGS(300-450℃),和(b)使用基于Cu的催化剂(例如Cu/ZnO/Al2O3)的低温WGS(200-270℃)。WGS发展的必要性是(i)产生氢用于实现哈伯 -博施法用于氨合成,(ii)用于去除一氧化碳,其充当系统如PEM燃料电池中的毒药。一氧化碳连同氢是用于烃和醇的费歇尔-特罗普希合成的有用前体。
如本文证实的,在本发明的方法中,在生物质的转化期间CO部分氧化为CO2。在过去,此类转化已使用多金属氧酸盐连同金催化剂一起进行。(30) 本发明基于下述发现:在矿物酸中单独的多金属氧酸盐H5PV2Mo10O40可用于在极低温度下进行水煤气变换反应。此类水煤气转化是目前实践的更高温水煤气变换反应的改良替代方案,因为(1)它在低温下在热力学上更有效 (ΔGr,g,25℃=-5.8kcal/摩尔),和(ii)分开的催化CO2和电化学H2形成步骤取消使CO2与H2分开的必要性。例如,在玻璃压力管中,使2巴CO与溶解于8mL 80%H2SO4水溶液中的1.4mmol H5PV2Mo10O40·36H2O反应。在100℃下5小时后,获得完全转化,并且H2如上所述电化学释放。
相应地,本发明的另一个实施例涉及用于由生物质或其组分或者碳水化合物制备氢(H2)的方法,该方法包括下述步骤:(a)在足以产生二氧化碳(CO2) 的条件下,在浓酸的存在下,使生物质、生物质组分或碳水化合物与多金属氧酸盐催化剂或其溶剂化物接触;和(b)电化学产生氢(H2);其中步骤(a)和(b) 同时或序贯执行。上文描述的反应条件中的任一种包括生物质类型、酸等可用于该工艺。每种可能性代表本发明的一个分开实施例。
对于通过包括水煤气变换反应的CO2产生,多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子的比率一般高于用于制备CO的相应工艺中。例如,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率优选大于1当量,例如约2当量或更大的多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。在一些实施例中,多金属氧酸盐与碳水化合物、生物质或生物质组分的比率为大于1至约2当量多金属氧酸盐/碳水化合物、生物质或生物质组分中的碳原子。上述条件优选用于依照本发明的一些实施例的水煤气变换反应。
水煤气变换反应的表示呈现于图2B中,其中CO的氧化连同最初贮存于多金属氧酸盐上的电子和质子的电化学氧化一起显示。
多金属氧酸盐催化剂
各种多金属氧酸盐催化剂可用于本发明的方法中。在一些实施例中,催化剂是由通式[XxMmOy]q-表示的可溶性多氧阴离子盐或其溶剂化物,其中X 是金属或非金属杂原子或者质子;M是选自钨(W)、钼(Mo)、铌(Nb)、钒(V)、钽(Ta)、铋(Bi)、锑(Sb)、锡(Sn)及其任何组合的增补原子;O是氧;x是0 至6的整数;m是4至200的整数;y是5至1000的整数;并且q是0至 30的整数。
多金属氧酸盐催化剂的一个非限制性类别是由通式Qq[XM12O40]表示的Keggin化合物或其溶剂化物,其中X选自(i)B、Al、Ga、In、Si、Ge、Sn、 P、As、Sb、S、Se、Te;(ii)质子;和(iii)选自Sc、Ti、V、Cr、Mn、Fe、 Co、Ni、Cu和Zn的过渡金属;M选自钨(W)、钼(Mo)及其组合,其中钨和 /或钼处于高价态,例如+4、+5或+6;Q是选自质子、碱金属、碱土金属、过渡金属包括镧系元素或锕系元素、主族金属、和有机阳离子例如季铵或鏻阳离子的抗衡阳离子;并且q是0至30的整数。Keggin结构具有基于中心 XO4四面体的近似四面体对称,所述中心XO4四面体由在四组三边缘共享的八面体M3O13中排列的十二个MO6八面体围绕。不希望受任何特定机制或理论束缚,可区分四个种类的氧原子:使杂原子与增补原子(addenda)连接的 4个内部氧、12个边缘共享氧、连接M3O13单位的12个转角共享氧和12个末端氧。
在一些实施例中,多金属氧酸盐催化剂由通式Qq[XM12-nM’nO40]表示或是其溶剂化物,其中Q、X、M和n如上定义;M’选自铌(Nb)、钽(Ta)、锑 (Sb)、铋(Bi)、锡(Sn)和钒(V);并且n是0、1、2、3、4、5或6。在其他实施例中,多金属氧酸盐是由式Qq[XMo12-nVnO40]表示的钒取代的钼酸盐。每种可能性代表本发明的一个分开实施例。在一个目前优选的实施例中,多金属氧酸盐催化剂是H5PV2Mo10O40。
多金属氧酸盐催化剂通常以溶剂化形式例如水合物发现。因此,本发明涵盖多金属氧酸盐溶剂化物例如但不限于多金属氧酸盐水合物的使用。多金属氧酸盐催化剂的其他溶剂化物分子包括但不限于二乙醚、乙腈、二甲亚砜、四氢呋喃、甲醇、乙醇溶剂化物等等。溶剂化物分子的量可从小于一到几百不等。每种可能性代表本发明的一个分开实施例。
目前优选的溶剂化形式是水合物。因此,本发明的多金属氧酸盐催化剂可采取半水合物、水合物、倍半水合物、二水合物、三水合物或多水合物的形式,其中水分子的数目可为高达几百。在一些实施例中,多金属氧酸盐催化剂是H5PV2Mo10O40的水合形式,例如H5PV2Mo10O40x nH2O,其中n是0 至36。一般地,水分子的数目范围可为约1/2至500个水分子。每种可能性代表本发明的一个分开实施例。在一个特定实施例中,多金属氧酸盐是 H5PV2Mo10O40x36H2O。
在本发明的方法中使用的多金属氧酸盐催化剂可以许多同分异构形式存在,其中每一种均由本发明涵盖。
对于本领域技术人员显而易见的是:本领域技术人员看起来适当的任何催化剂与碳水化合物的比率均可用于本发明的上下文中。
生物质
如本文使用的,术语“生物质”指来源于活生物体或近期活的生物体的生物材料。生物质优选陆生生物质。生物质仍是迄今为止最大的生物质能源;例子包括森林残留物(例如枯死的树木、树枝和树桩)、庭院剪枝、木屑和甚至城市固体或农业废弃物。生物质可由众多类型的植物产生,包括但不限于芒草、柳枝稷、大麻、玉米、白杨、柳树、高粱、甘蔗(sugarcane)、竹、甘蔗(sugar cane)、甘蔗渣、棉花,以及各种树种,例如松、榉木、核桃、桉树、和棕榈油。生物质还可由秸秆产生,所述秸秆是农业副产物,在已去除谷粒和谷壳后的谷类植物的干燥茎。秸秆构成谷类作物例如大麦、燕麦、稻、黑麦和小麦约一半的得率。生物质还可由卡纸板产生。上文描述的任何生物质源以及其他生物质源可用于根据本发明的原理产生CO和H2,其中每种可能性代表本发明的一个分开实施例。
通常,陆生生物质含有35–50%纤维素、20–35%半纤维素和10–25%木质素。
如本文使用的,术语“纤维素”指由几百个至超过一万个β(1→4)连接的 D-葡萄糖单位(C6H12O6)n的线性链组成的多糖,是绿色植物、许多形式的藻类和卵菌纲的初生细胞壁的重要结构组分。在该术语中包括的是所有已知形式的纤维素例如粉末纤维素和微晶纤维素。
如本文使用的,术语“半纤维素”指连同纤维素一起存在于几乎所有植物细胞壁中的几种杂聚物(基质多糖)中的任一种。半纤维素包括木聚糖、葡糖醛酸木聚糖、阿拉伯木聚糖、葡甘露聚糖和木糖葡聚糖。虽然纤维素仅含有无水葡萄糖,但半纤维素中的单体可包括木糖、甘露糖、半乳糖、鼠李糖和阿拉伯糖。半纤维素主要含有D-戊糖以及偶然的少量L-糖。
如本文使用的,术语“木质素”指通过生物化学来源于氨基酸苯丙氨酸的对羟基肉桂醇的氧化偶联形成的高度交联的羟基化和甲氧基化苯丙烷聚合物。木质素是植物的次生细胞壁的整合部分。
碳水化合物
如本文使用的,术语“碳水化合物”指包含碳、氢和氧的有机化合物,通常具有经验式Cm(H2O)n(其中m与n相同或不同)。术语碳水化合物与术语“糖类”同义,所述“糖类”包括单糖、二糖、三糖、寡糖和多糖。碳水化合物可来源于植物生物质或者任何其他天然或合成源。
“单糖”指多羟基醛(醛糖)或多羟基酮(酮糖)及其衍生物和类似物。单糖可为D-或L-构型。单糖可为吡喃糖(6元环)或呋喃糖(5元环)形式中的D-或L-环状糖形式。此外,单糖包括5碳糖(戊糖)或6碳糖(己糖)。单糖的例子包括但不限于阿洛糖、阿卓糖、阿拉伯糖、克拉定糖、赤藓糖、赤藓酮糖、果糖、岩藻糖、墨角藻糖、半乳糖、葡萄糖、葡萄糖-6-磷酸、古洛糖甘油醛、L-甘油-D-甘露-庚糖、甘油酮、古洛糖、艾杜糖、来苏糖、甘露糖、甘露糖-6-磷酸、阿洛酮糖、奎诺糖、鼠李糖、核糖、核酮糖、景天庚酮糖、山梨糖、塔格糖、塔罗糖、苏糖、木糖和木酮糖。单糖还可为脱氧糖 (醇羟基替换为氢)、C-取代单糖(在非末端碳原子处的氢替换为碳)、不饱和单糖、醛糖醇或糖醇(羰基替换为CHOH基)(例如山梨糖醇、乙二醇、甘油、赤藓糖醇、苏糖醇、阿糖醇、木糖醇、核糖醇、甘露醇、半乳糖醇 (galacitol)、岩藻糖醇、艾杜糖醇、肌醇、庚七醇、异麦芽糖(osomalt)、麦芽糖醇、乳糖醇、麦芽三糖醇、麦芽四糖醇、聚甘油(polyglycitol)、鼠李糖醇等)、醛糖酸(醛基替换为羧基)(例如葡糖酸)、酮醛糖酸、糖醛酸、醛糖二酸(例如酒石酸)等等。
单糖可连接在一起以形成二糖、三糖、寡糖或多糖。“多糖”指由通过半缩醛或糖苷键彼此邻接的约500至超过100,000个糖单位形成的聚合物。术语“二糖”、“三糖”和“多糖”包括但不限于阿比可糖、阿卡波糖、amicetose、支链淀粉、直链淀粉、芹菜糖、阿卡波糖、蛔糖、抗坏血酸、波伊文糖、纤维二糖、纤维三糖、纤维素、马铃薯三糖、查耳糖、壳多糖、可立糖、环糊精、磁麻糖、糊精、2-脱氧核糖、2-脱氧葡萄糖、2-脱氧毛地黄糖、毛地黄糖、洋地黄毒糖、evalose、evemitrose、果寡糖、半乳寡糖、龙胆三糖、龙胆二糖、葡聚糖、糖原、肝糖、金缕梅糖、肝素、菊粉、异左旋葡萄糖酮 (isolevoglucosenone)、异麦芽糖、异麦芽三糖、异葡糖基麦芽糖、曲二糖、乳糖、昆布二糖(laminarabiose)、左旋葡聚糖、左旋葡萄糖酮、β-麦芽糖、麦芽三糖、甘露寡糖、甘露三糖、松三糖、蜜二糖、胞壁酸、霉菌糖、6-脱氧 -D-阿洛糖、神经氨酸、黑曲霉糖、野尻霉素、诺维糖、齐墩果糖、潘糖、泊雷糖、车前糖、樱草糖、棉子糖、玫红糖、芸香糖、箭毒羊角拗糖、景天庚酮糖、景天庚醛聚糖、茄三糖、槐糖、水苏糖、链霉糖、蔗糖、α-海藻糖、松二糖、泰威糖、木二糖、木聚糖、伞形糖、淀粉和淀粉衍生物等等。
在一个实施例中,碳水化合物是来源于陆生生物质的多糖,即如上所述的纤维素、半纤维素或其组合。
如本文和所附权利要求中使用的,单数形式“一个”、“一种”和“该/所述”包括复数指代,除非上下文另有说明。
本文引用的所有参考文献均以引用的方式在此全文并入,如同它在本文中完全阐述一样。
本发明的原理借助于下述非限制性实例加以证实。
实验细节部分
实例1:在有氧条件下的葡萄糖转化
基于由一般而言的H5PVV2Mo10O40(22,23)以及特别的醇类和邻位醇(15) 催化的电子转移-氧转移反应的机械分析,考虑根据方案2中呈现的一系列反应的D-葡萄糖氧化可获得5当量HCOOH、1当量HCHO和6当量的还原型多金属氧酸盐H7PVIV2Mo10O40。
方案2.D-葡萄糖的计划氧化切割,为了简单起见通过六当量 H5PVV2Mo10O40作为费舍尔投影呈现。
不希望受任何特定理论或作用机制束缚,推测经由来自多金属氧酸盐的氧原子插入伴随碳-碳键切割理想地进行,以获得来自C-1至C-5碳原子的甲酸(HCOOH)和来自C-6碳原子的甲醛(HCHO)。脱氧合和还原型多金属氧酸盐H5PVIV2Mo10O39与H2O反应,以获得H7PVIV2Mo10O40。如果反应在O2的存在下进行,则它与H7PVIV2Mo10O40的反应获得H5PVV2Mo10O40和H2O。 2个电子和2个质子或氧化的H2/多金属氧酸盐单位H7PVIV2Mo10O40将会丧失或消耗。
使用甲醇/水作为溶剂测试如方案2中提出的D-葡萄糖氧化,与文献报道(16,17)相比较,其允许在相对温和的条件下反应,尤其是不需要高O2压。因此,100mg(0.56mmol)D-葡萄糖和80mg(0.038mmol) H5PV2Mo10O40·36H2O溶解于甲醇和水的3mL(1:1)混合物中,并且将溶液在 2巴O2下加热至110℃共18小时。在这些条件下,根据方案3获得D-葡萄糖的基本定量转化,其中在酸性反应介质中的甲醇的存在导致进一步的部分酯、半缩醛和缩醛形成。应注意到甲醇在这些条件下仅以痕量被氧化。
方案3
如可见的,结果证实与方案2中呈现的预期理论结果的良好关联。
实例2.在有氧条件下的碳水化合物的转化
其他碳水化合物的转化通过100mg反应和80mg H5PV2Mo10O40在3mL (1:1)甲醇/水中在2巴O2下在110℃下进行18小时。作为每反应底物的碳原子给出的产物量在表1中给出。
表1.各种糖类和多糖的有氧氧化。
nd–未检测到。[a]30小时。[b]120℃,30小时。
实例3.在浓H2SO4中的碳水化合物至CO的转化
根据方案4,纤维素或半纤维素可转化为CO和还原型多金属氧酸盐。所使用的纤维素是来自天然木浆的微晶粉末,并且半纤维素是来自榉木的木聚糖。两者均不经任何预处理而使用。反应通过将100mg多糖和7.80g H5PV2Mo10O40在8mL浓H2SO4中在70℃下在N2气氛下混合5小时来进行。因为多金属氧酸盐被36个水分子溶剂化,所以在反应期间的H2SO4浓度为~80%。存在关于纤维素和半纤维素的完全转化,并且通过用热导检测器 (GC-TCD)的气相色谱分析气相显示仅形成CO和CO2,其比率经过五次实验平均为65±3:35±3。另外,使用GC-TCD伴随N2作为内部标准的定量分析显示定量形成,即97±4%的CO和CO2。
H5PV2Mo10O40的还原程度通过用KMnO4滴定进行测定。经过五次纤维素氧化实验求平均值,测量2.7±0.2当量电子/当量碳原子。因为钒在钼之前还原,(22,23)所以通过反应化学计量学,H7PVIV2MoVI10O40和 H8PVIV2MoVMoVI9O40明显以~3:7比率存在于反应混合物中。
方案4.使用H5PV2Mo10O40的纤维素和木聚糖半纤维素的无氧氧化
实例4.CO的转化
在实例3中呈现的条件下的CO2形成原因并未立即明确。根据实例3的反应证实在仅30分钟后,CO:CO2比率为5:1,并且因此考虑CO通过 H5PV2Mo10O40被氧化。事实上,~2mmol CO对H5PV2Mo10O40在H2SO4中的 0.42M溶液的添加显示在70℃下5小时后伴随~40%转化的CO2形成(方案 5):
CO+H5PVV2Mo10O40+H2O→CO2+H7PVIV2Mo10O40
方案5.CO的氧化
实例5.在各种溶剂中的纤维素至CO的转化
在各种酸溶剂中的纤维素转化通过将100mg纤维素和7.80g H5PV2Mo10O40在8mL溶剂中在N2下混合来进行,如表2中呈现的。
表2.使用不同酸溶剂的纤维素的无氧氧化。
反应条件:100mg多糖,7.80g(3.3mmol)H5PV2Mo10O40·36H2O,8mL 溶剂,70,℃5小时,N2。因为多金属氧酸盐由H2O溶剂化,所以在反应混合物中还另外存在1.9mL H2O。
实例6.H2的释放
图1A,这种反应溶液,即在H2SO4中的0.42M H5PV2Mo10O40,由2.7 个电子还原的循环伏安法测量显示0.4V相对于NHE的氧化还原电势。在0.6V下的25-30%多金属氧酸盐氧化导致溶液变色为暗绿色,以及氧化还原电位至0.05V相对于NHE的变换,图1B。根据这点,看起来 H8PVIV2MoVMoVI9O40首先氧化为H7PVIV2MoVI10O40,其随后氧化为 H5PVV2Mo10O40。因此,为了定量形成H2,使用Pt片电极氧化溶液。对于所需库仑总数目的25-30%,氧化在0.6V下,并且随后多金属氧酸盐的氧化在 0.2V下完成。总库仑计数获得与KMnO4滴定相同的平均电子量。收集氢并且通过GC-TCD验证其定量形成。
用于实例1-6的材料与方法
材料。微晶纤维素、半纤维素及其他糖购自Sigma-Aldrich。微晶纤维素不经预处理而使用。H5PV2Mo10O40·36H2O通过已知操作进行制备。(25)
方法。
(1)在水/甲醇中的有氧反应:将100mg糖类或多糖、80mg H5PV2Mo10O40·36H2O(0.038mmol)溶解于CD3OD和D2O在50mL玻璃压力管中的3mL(1:1)混合物中。反应在2巴O2、110℃下进行18小时。使用300 MHz 1H NMR和乙醇作为外部标准计算所形成的产物:甲酸、甲酸甲酯、二甲氧基甲烷、甲氧基甲醇和甲烷二醇的量。因为H2O的峰(始终在一定程度上存在)与甲醛衍生物的那些部分重叠,所以加入2滴35%HCl,以将水的峰偏移向低场。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8"不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD测定 CO2和CO。载气为Ar,柱T=120℃。CO2的保留时间–4.5分钟;CO的保留时间–30.2分钟。
(2)浓(98%)硫酸中的无氧氧化和氢形成:将100mg纤维素或半纤维素和 7.80g(3.3mmol)H5PV2Mo10O40在8mL H2SO4中混合。在室温下,这是浆料,但在加热后,获得澄清溶液。反应在70℃下在N2下进行5小时。如上所述分析CO和CO2。使用铂片、在0.7V下的25-30%氧化和在0.2V下的剩余部分,电化学形成H2。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8"不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD 定量H2。载气为Ar,柱T=120。℃保留时间–10分钟。
(3)在各种酸溶剂中的纤维素的无氧氧化:将100mg纤维素和7.80g(3.3 mmol)H5PV2Mo10O40在8mL溶剂中混合。反应在70℃下执行5小时。
(4)循环伏安法实验:使用与个人计算机连接的恒电位仪(CHI660A)执行这些实验。测量在由下述组成的三电极电池配置中执行:(i)铂片(工作电极)、 (ii)Pt线(对电极)和(iii)Pt(参考电极)。实验在室温下执行。扫描速率为 200mV/秒。
(5)用KMnO4滴定:通过用标准草酸溶液的体积滴定测定新鲜KMnO4溶液的浓度。使用还原型多金属氧酸盐在750nm处的峰的消失作为指示剂,体积滴定还原型多金属氧酸盐溶液。
实例1-6的结论
总之,基于前述实验,可得出下述结论:
(1)本发明提供了浓硫酸(廉价的工业商品)在多金属氧酸盐催化的反应中的首次报道的使用,所述多金属氧酸盐催化的反应目前允许纤维素和半纤维素两者以及其他生物质和生物质组分在温和反应温度下的完全转化。最初形成的甲酸以高得率形成,并且随后定量脱水为CO。在相对低的0.2V超电势下,关于大多数部分的H2的电化学释放允许合成气体,1当量CO和H2/ 碳原子的总体形成。
(2)H2SO4的使用具有附加优点:它显著增加电子转移-氧转移氧化速率,并且是在低电位下的H2释放的方便介质。
(3)通过净化反应混合物快速去除形成的CO可阻止其进一步氧化为 CO2。然而,如果H2是靶,则可使用通过使用这种催化系统的水煤气变换反应的CO的完全转化。以这种方式,可形成2当量H2/多糖中的碳原子(参见实例7)。
(4)H2的分开电化学释放可允许H2和CO的分开形成。这些可依照本发明的一些实施例组合成合成气。可替代地,如果需要分开的H2形成,则它可不依赖于CO而生成。
(5)重要的是,在仅气态产物形成后,反应溶液即在H2SO4中的 H5PV2Mo10O40可再循环且再使用,而无需任何另外的处理。使用纤维素作为底物的十个反应循环显示反应性中无变化。
实例7.水煤气变换反应
水煤气变换(WGS)反应在如表3中所示的环境条件下进行。
表3.在不同多金属氧酸盐上进行的WGS反应
反应条件:所有反应均在具有2atm CO、3g多金属氧酸盐(1.38mmol)、 8mL 80%硫酸的Fisher-Portar压力管中进行。(a)转化基于所使用的多金属氧酸盐。使用铂片、在0.7V下的25-30%氧化和在0.2V下的剩余部分,电化学形成H2。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8"不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD定量H2。载气为Ar,柱T=120℃。保留时间–10分钟。
实例8-11.生物质(麦秸)的转化
根据表4中所示的量和条件,使磨碎的麦秸与在5mL 80%H2SO4中的 H5PV2Mo10O40在1巴N2下在15mL玻璃管中反应。结果也在表3中给出。通过使用铂工作电极、参考电极和对电极在0.5V下的电解回收H2。可替代地,形成1当量H2/当量CO,并且形成2当量H2/当量CO2。
表4.
得率计算为相对于根据PV=nRT转变为摩尔的1巴N2以摩尔计的形成的总CO和CO2。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8" 不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD测定 CO2和CO。载气为Ar,柱T=120℃。CO2的保留时间–4.5分钟;CO的保留时间–30.2分钟。
实例12-15.各种生物质的转化
使磨碎的生物质样品(20mg)与在5mL 80%H2SO4中的2g H5PV2Mo10O40在60℃下在1巴N2下在15mL玻璃管中反应4小时。结果在表5中给出。通过使用铂工作电极、参考电极和对电极在0.5V下的电解回收H2。大约形成1当量H2/当量CO,并且形成2当量H2/当量CO2。
表5.
得率计算为相对于根据PV=nRT转变为摩尔的1巴N2以摩尔计的形成的总CO和CO2。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8" 不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD测定 CO2和CO。载气为Ar,柱T=120℃。CO2的保留时间–4.5分钟;CO的保留时间–30.2分钟。
实例16-19.木质素的转化。
根据表5中所示的量和条件,使木质素(来自TCI的碱性)与在5mL 80% H2SO4中的H5PV2Mo10O40在1巴N2下在15mL玻璃管中反应。结果也在表 6中给出。通过使用铂工作电极、参考电极和对电极在0.5V下的电解回收 H2。大约形成1当量H2/当量CO,并且形成2当量H2/当量CO2。
表6.
CO和CO2的得率计算为相对于根据PV=nRT转变为摩尔的1巴N2以摩尔计形成的。使用GowMac仪器,使用与填充有HayeSep T的4'x 1/8"不锈钢柱串联的填充有分子筛的20'x 1/8"不锈钢柱,通过GC-TCD测定 CO2和CO。载气为Ar,柱T=120℃。CO2的保留时间–4.5分钟;CO的保留时间–30.2分钟。
虽然本发明的某些实施例已得到举例说明和描述,但明确的是本发明并不限于本文描述的实施例。众多修饰、变化、变动、置换和等价物对于本领域技术人员是显而易见的,而不背离如由下述权利要求所述的本发明的精神和范围。
参考文献:
1.C.-H.Zhou,X.Xia,C.-X.Lin,D.-S.Tong,J.Beltramini,Catalytic conversion of lignocellulosic biomass to fine chemicals and fuels.Chem.Soc. Rev.40,5588-5617(2011).
2.J.A.Melero,J.Iglesias,A.Garcia,Biomass as renewable feedstock in standard refinery units.Feasibility,opportunities and challenges.Energy Environ. Sci.5,7393-7420(2012).
3.L.T.Fan,M.M.Gharpuray,Y.H.Lee,Cellulose Hydrolysis (Springer-Verlag,Berlin,1987).
4.L.Zhu,J.P.O’Dwyer,V.S.Chang,C.B.Brabda,M.Y.Holtzapple, Structural features affecting biomass enzymatic digestibility.Bioresource Tech. 99,3817-3828(2008).
5.J.N.Chheda,G.W.Huber,J.A.Dumesic,Liquid-phase catalytic processing of biomass-derived oxygenated hydrocarbons to fuels and chemicals. Angew.Chem.Int.Ed.46,7164-7183(2007).
6.G.W.Huber,S.Iborra,A.Corma,Synthesis of transportation fuels from biomass:chemistry,catalysts,and engineering.Chem.Rev.106,4044-4098 (2006).
7.E.L.Kunkes,D.A.Simonetti,R.M.West,J.C.Serrano-Ruiz,C.A. J.A.Dumesic,Catalytic conversion of biomass to monofunctional hydrocarbons and targeted liquid-fuel classes.Science,322,417-421(2008).
8.G.W.Huber,J.N.Chheda,C.J.Barrett,J.A.Dumesic,Production of liquid alkanes by aqueous-phase processing of biomass-derived carbohydrates. Science,308,1446-1449(2005).
9.A.D.Sutton,F.D.Waldie,R.Wu,M.Schlaf,L.A.‘Pete’Silks III,J.C. Gordon,The hydrodeoxygenation of bioderived furans into alkanes.Nature Chem. 5,428-432(2013).
10.G.W.Huber,J.W.Shabaker,J.A.Dumesic,Raney Ni-Sn catalyst for H2production from biomass-derived hydrocarbons.Science,300,2075-2077 (2003).
11.T.D.Matson,K.Barta,A.V.Iretskii,Peter C.Ford,One-pot catalytic conversion of cellulose and of woody biomass solids to liquid fuels.J.Am.Chem. Soc.133,14090-14097(2011).
12.J.B.Binder,R.T.Raines,Simple chemical transformation of lignocellulosic biomass into furans for fuels and chemicals.J.Am.Chem.Soc. 131,1979-1985(2009).
13.P.M.Maitlis,A.de Klerk,Greener-Fischer-Tropsch-Processes for Fuels and Feedstocks,Wiley-VCH,Weinheim,2013
14.A.B.Stiles,Methanol,past,present,and speculation on the future. AIChE,Journal,23,362-375,(1977).
15.A.M.Khenkin,R.Neumann,Oxidative C-C bond cleavage of primary alcohols and vicinal diols catalyzed by H5PV2Mo10O40 by an electron transfer and oxygen transfer mechanism.J.Am.Chem.Soc.130,14474-14476 (2008).
16.J.Albert,R.A.P.Wasserscheid.Selective oxidation of complex,water-insoluble biomass to formic acid using additives as reaction accelerators,Energy Environ.Sci.5,7956-7962(2012).
17.R.N.Taccardi,A.P.Wasserscheid,Selective catalytic conversion of biobased carbohydrates to formic acid using molecular oxygen.Green Chem.11,2759-2763,(2011).
18.J.Li,D.-J.Ding,L.Deng,Q.-X.Guo,Y.Fu,Catalytic air oxidation of biomass-derived carbohydrates to formic acid.ChemSusChem,5,1313-1318 (2012).
19.C.Fellay,P.J.Dyson,G.Laurenczy,A viable hydrogen-storage system based on selective formic acid decomposition with a ruthenium catalyst. Angew.Chem.Int.Ed.47,3966–3968(2008).
20.Z.-L.Wang,J.-M.Yan,Y.Ping,H.-L.Wang,W.-T.Zheng,Q.Jiang, An efficient CoAuPd/C catalyst for hydrogen generation from formic acid at room temperature.Angew.Chem.Int.Ed.52,4406-4409(2103).
21.J.M.Trillo,G.Munuera,J.M.Criado,Catalytic decomposition of formic acid on metal oxides.Catal.Rev.Sci.Eng.7,51-86(1972).
22.R.Neumann,Activation of molecular oxygen,polyoxometalates and liquid phase catalytic oxidation.Inorg.Chem.49,3594-3601(2010).
23.I.Efremenko,R.Neumann,Computational insight into the initial steps of the Mars-van Krevelen mechanism:Electron transfer and surface defects in the reduction of oolyoxometalates.J.Am.Chem.Soc.134,20669-20680 (2012).
24.G.Jacobs,B.H.Davis,Low temperature water-gas shift catalysts. Catalysis 20,122-285(2007).
25.G.A.Tsigdinos,C.J.Hallada,Molybdovanadophosphoric acids and their salts.I.Investigation of methods of preparation and characterization.Inorg. Chem.7,437-441(1968).
26.I.A.Weinstock,R.H.Atalla,R.S.Reiner,M.A.Moen,K.E.Hammel, C.J.Houtman,C.L.Hill,M.K.Harrup Journal of Molecular Catalysis A: Chemical 116,59-84,(1997).
27.David S.Newsome(1980)The Water-Gas Shift Reaction,Catalysis Reviews:Science and Engineering,21:2,275-318.
28.Bartholomew,C.H.;Farrauto,R.J.Fundamentals of Industrial Catalytic Processes,2nd ed.;John Wiley and Sons:2006.
29.H.P.Dhar,L.G.Christner,and A.K.Kush;J.Electrochem.Soc. 1987 134(12),3021-3026.
30.W.B.Kim,T.Voitl,G.J.Rodriguez-Rivera,J.A.Dumesic, Powering fuel cells with CO via aqueous polyoxometalates and gold catalysts. Science,305,1280-1283(2004).
- 还没有人留言评论。精彩留言会获得点赞!