一种改性黑磷烯及其制备方法与应用
1.本发明涉及新型二维材料黑磷烯制备和应用技术领域,尤其涉及一种改性黑磷烯及其制备方法与应用。
背景技术:
2.2014年,一种新型二维材料单层黑磷—黑磷烯(pn)通过胶带技术被制备出来,与石墨烯相比,黑磷烯的最大理论表面积为2400m2g-1
,具有更高的导电(载流子迁移率达到160000cm2/(v
·
s))和导热性能(导热系数为8500w/(m
·
k)),同时,黑磷烯可以和o2反应释放出大量热,相比于石墨烯能量惰性,黑磷烯可以作为一种含能组分。但是,黑磷烯的应用过程还面临一个非常大的难题,即它在空气中容易降解,其原因是:黑磷烯在平面外具有很强的偶极矩,使得黑磷烯表面易吸附水和氧生成磷氧化合物从而降解。为扩大黑磷烯的应用领域,已经开始研究黑磷烯的稳定性,但研究结果并不理想,其原因是:在空气中稳定黑磷烯的同时,会引进一定量的其他元素原子或基团,从而改变黑磷烯的物理化学性质,进而影响其实际的应用。尤其是将黑磷烯引入到含能复合材料时,会因为新元素或基团的引入而导致体系能量水平下降。
3.另外,有研究学者将黑磷烯纳米片作为二维纳米填料用于增强聚乙烯醇(pva)基体时发现,当加入量为3.11wt%,达到最大拉伸强度316.9mpa,是纯pva的的1.9倍,随着加入量的进一步增加,机械性能开始下降。导致此结果的原因是黑磷烯纳米片出现了团聚现象。
4.因此,如何解决黑磷烯在空气中易氧化以及黑磷烯作为二维填料增强基体时易出现团聚的现象成为了本领域技术人员亟需解决的问题。
技术实现要素:
5.有鉴于此,本发明提供了一种改性黑磷烯及其制备方法与应用,有效解决了黑磷烯在空气中易氧化以及黑磷烯作为二维填料增强基体时易出现团聚的现象等技术问题。
6.为了达到上述目的,本发明采用如下技术方案:
7.本发明提供了一种改性黑磷烯的制备方法,包括以下步骤:
8.s1、将黑磷晶体制备成bp-水溶液,再将bp-水溶液制备成黑磷烯;
9.s2、将步骤s1得到的黑磷烯分散在反应溶剂中得到黑磷烯分散液,再向黑磷烯分散液中加入聚叠氮化合物得到黑磷烯混合液;
10.s3、将步骤s2得到的黑磷烯混合液进行热处理,生成改性黑磷烯;
11.以上步骤均在保护气体下进行。
12.所述步骤s1中,bp-水溶液的浓度为1~10mg/ml。
13.所述步骤s2中,黑磷烯与反应溶剂的溶度比为1~5mg/ml,黑磷烯与聚叠氮化合物的质量比为1:50~70;
14.所述反应溶剂为n,n-二甲基甲酰胺、二甲基亚砜、n-甲基吡咯烷酮、聚乙二醇中的
一种或几种。
15.所述步骤s2中,所述聚叠氮化合物为聚叠氮缩水甘油醚、聚3-叠氮甲基-3,甲基氧杂环丁烷、聚3,3-双叠氮甲基氧杂环丁烷、3,3-双叠氮甲基环氧丁烷-四氢呋喃共聚醚中的一种或几种。
16.所述步骤s2中,加入聚叠氮化合物后进行搅拌,得到黑磷烯混合液;所述搅拌速度为100~1500rpm,搅拌时间为1~5h。
17.所述的保护气体为氮气、氩气、氦气、氖气和二氧化碳中的一种。
18.所述步骤s3中,进行热处理时,以2~10℃/min速率升温至100~200℃,然后保温12~60h。
19.对热处理后的混合物再进行冷却、离心、洗涤、干燥即得改性黑磷烯;
20.所述冷却温度为室温;
21.所述离心的时间为10~30min,离心的转速为3000~10000rpm;
22.所述洗涤时先用反应溶剂进行洗涤,再用水或乙醇进行洗涤;
23.所述干燥的方式为介质为二氧化碳的临界点干燥、超临界干燥、真空干燥和冷冻干燥中的一种。
24.本发明提供了由上述方法所制备的改性黑磷烯。
25.本发明还提供了一种上述改性黑磷烯在催化剂材料、电化学材料或航空材料中的应用。
26.本发明所述技术方案的原理为:pn表面p元素与叠氮聚合物的叠氮基形成p=n双键,占据p元素表面的孤对电子,实现磷原子的五配位键合,从而防止孤对电子与空气中的水和氧反应,使pns完全钝化。
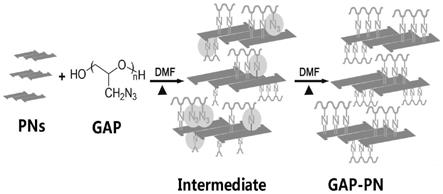
27.经由上述的技术方案可知,与现有技术相比,本发明的有益效果如下:
28.1、本发明方法制备的黑磷烯具有在空气中稳定,不被氧化降解的特点。
29.2、本发明方法采用聚叠氮化合物作为黑磷烯稳定修饰剂,将黑磷烯引入到含能复合材料时,不会导致体系能量水平下降。
30.3、本发明方法采用聚叠氮化合物作为黑磷烯稳定修饰剂,引入了带有活性羟基基团,可以参加聚氨酯等可以和羟基反应的体系,提高界面作用力,促进相容性,降低团聚现象,提高体系的机械性能。
31.4、本发明制备过程简单可控。
32.5、由于本发明使用的处理、干燥设备简单常见,不需要添加昂贵设备,,因此属于低成本实验绿色制备工艺。
33.6、本发明利用聚叠氮化合物的叠氮基与黑磷烯表面反应生成p=n基团,可以增大黑磷烯在空气中的稳定性,并且,所制备的改性黑磷烯还具有黑磷纳米片本身的良好的特性:良好的载流子迁移率、可调谐的带隙和高度的各向异性等,因而本发明所制备的改性黑磷烯可广泛应用于催化剂材料、电化学材料或航空材料等领域。
附图说明
34.图1中,a图和b图分别是实施例1得到的pns的tem和hr-tem图像,c图和d图分别是实施例1得到的gap-pn的tem和hr-tem图像,e图为gap-pn的stem图像和stem-edx元素映射
图像;
35.图2是gap功能化pns的反应过程图;
36.图3中,(a)图和(b)图分别是实施例1得到的pns的3dafm图像和afm图像,(c)图和(d)图分别是实施例1得到的gap-pn的3dafm图像和afm图像,(e)图和(f)图分别是实施例1得到的pns和gap-pn的高度图;
37.图4中,(a)图为实施例1得到的pns和gap-pn的拉曼光谱图,(b)图为原始pns和gap-pn的
31
p-nmr谱图(*为自旋边带峰),(c)图和(d)图分别为原始pns和gap的xps光谱图,(e)图和(f)图均为实施例1得到的gap-pn的xps光谱图;
38.图5是pn表面p元素与gap叠氮基形成p=n双键的反应机理图;
39.图6中,(a)图和(c)图分别为实施例1得到的pns和gap-pn-48h的xps光谱图,(b)图和(d)图分别为实施例1得到的在空气中放置30天后的pns和gap-pn-48h的xps光谱图;(e-g)图为实施例1得到的pns、gap-pn-12h和gap-pn-48h分散在di水中并放置在空气中1、7、14、21、28、35、49和63天的紫外/可见吸收光谱图;(h)图为pns、gap-pn-12h和gap-pn-48h的降解比(1-a/a0);
40.图7中,(a)图为实施例1得到的pns、gap-pn和gap的dsc图(10℃/min),(b)图为实施例1得到的pns和gap-pn分解过程中释放的热量。
具体实施方式
41.本发明提供了一种改性黑磷烯的制备方法,包括以下步骤:
42.s1、将黑磷晶体制备成bp-水溶液,再将bp-水溶液制备成黑磷烯;
43.s2、将步骤s1得到的黑磷烯分散在反应溶剂中得到黑磷烯分散液,再向黑磷烯分散液中加入聚叠氮化合物得到黑磷烯混合液;
44.s3、将步骤s2得到的黑磷烯混合液进行热处理,生成改性黑磷烯;
45.以上步骤均在保护气体下进行。
46.在本发明的步骤s1中,bp-水溶液的浓度为1~10mg/ml,优选为1~7mg/ml。
47.优选地,将bp-水溶液加入烧杯置于冰浴中,放入细胞破碎仪,在保护气体氛围下,样品底部距离超声探头1.5~3cm,超声功率为45~95%,以超声开2s、关4s的模式工作2~30h,再经过离心、冷冻干燥得到黑磷烯;
48.进一步优选,超声工作8~20h,超声功率为45~80%。
49.优选地,离心时,转速为5000~10000rpm,离心时间为5~20min;冷冻干燥时的冷冻温度为-50~-10℃,时间为8~12h;
50.进一步优选,离心时,转速为7000~10000rpm,离心时间为10~15min;冷冻干燥时的冷冻温度为-50~-30℃,时间为10~12h。
51.在本发明的步骤s2中,将步骤s1得到的黑磷烯超声分散在反应的溶剂中,超声功率45~95%,时间30~120分钟;然后向黑磷烯的分散液中加入聚叠氮化合物,100~1500rpm机械搅拌处理1~5小时,制得均匀的黑磷烯混合液。
52.优选地,超声功率为60~95%,超声时间为30~60分钟。
53.优选地,黑磷烯与反应溶剂的溶度比为3~5mg/ml,黑磷烯与聚叠氮化合物的质量比为1:50~60;
54.进一步优选,黑磷烯与反应溶剂的溶度比为4mg/ml,黑磷烯与聚叠氮化合物的质量比为1:60;
55.优选地,反应溶剂为n,n-二甲基甲酰胺。
56.优选地,聚叠氮化合物为聚叠氮缩水甘油醚。
57.优选地,向黑磷烯的分散液中加入聚叠氮化合物,250~1000rpm机械搅拌处理1~3小时。
58.在本发明的步骤s3中,将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入保护气体,重复3次,然后以2~10℃/min速率升温至100~200℃,保温12~60h,冷却后,将混合物以3000~10000rpm离心10~30min,然后先用反应溶剂洗涤,再去离子水进行洗涤,各洗涤3~10次,再将其干燥3~10小时即得改性黑磷烯。
59.优选地,进行热处理时,以5~7℃/min速率升温至150~200℃。
60.优选地,进行离心时,离心的时间为10~20min,离心的转速为5000~7000rpm。
61.优选地,反应溶剂和去离子水各洗涤3~7次,再将其干燥3~7小时。
62.下面将对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
63.实施例1
64.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为1mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氩气氛围下,样品底部距离超声探头2cm,超声功率为45%,以超声开2s、关4s的模式工作8h,再经过7000rpm离心10min、-50℃下冷冻干燥10h得到黑磷烯;
65.s2、将步骤s1得到的黑磷烯(50mg)超声分散在n,n-二甲基甲酰胺(dmf,10ml)中,超声功率95%,时间30分钟;然后向黑磷烯的分散液中加入聚叠氮缩水甘油醚(gap,3g),250rpm机械搅拌处理1小时,制得均匀的黑磷烯混合液,整个过程在氩气下进行。
66.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氩气,重复3次,然后以5℃/min速率升温至150℃,保温12h,冷却后,将混合物以7000rpm离心10min,然后再用n,n-二甲基甲酰胺和去离子水各洗涤3次,再将其干燥3小时即得改性黑磷烯。
67.由本发明的图1可知,pns和gap-pn具有典型的多层结构,gap-pn表面上可以观察到黑色的物质,为pns表面上接枝的gap聚合物;另外,gap-pn的层间距离扩展了36%(从到),说明gap聚合物插入pns]的层间,使层间距离变大;tem-edx测试表明,磷(p)、氮(n)、碳(c)、氧(o)元素均匀分散在整个纳米片中。
68.由本发明的图3可知,pns和gap-pn的厚对相对平均,划线处pns的高度为4.37nm,gap-pn的高度为10.4nm。
69.由本发明的图4可知,合成了一种新型的共价功能化含p=n双键的黑磷纳米片(gap-pn)。
70.由本发明的图6可知,本发明制备的gap-pn具有良好的稳定性,在空气中放置60天后,pns和gap-pn-48h降解率分别为54.9%和8.8%。
71.由本发明的图7可知,与pns相比,gap-pn的放热峰值温度(t
p
)降低44.6℃,分解过程中放热量高达3154.9j/g,是pns的6.09倍。
72.实施例2
73.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为1mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氮气氛围下,样品底部距离超声探头2cm,超声功率为45%,以超声开2s、关4s的模式工作8h,再经过7000rpm离心10min、-50℃下冷冻干燥10h得到黑磷烯;
74.s2、将步骤s1得到的黑磷烯(50mg)超声分散在n,n-二甲基甲酰胺(dmf,10ml)中,超声功率95%,时间30分钟;然后向黑磷烯的分散液中加入聚叠氮缩水甘油醚(gap,3g),250rpm机械搅拌处理1小时,制得均匀的黑磷烯混合液,整个过程在氮气下进行。
75.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氮气,重复3次,然后以5℃/min速率升温至150℃,保温24h,冷却后,将混合物以7000rpm离心10min,然后再用n,n-二甲基甲酰胺和去离子水各洗涤3次,再将其干燥3小时即得改性黑磷烯。
76.实施例3
77.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为1mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氦气氛围下,样品底部距离超声探头2cm,超声功率为45%,以超声开2s、关4s的模式工作8h,再经过7000rpm离心10min、-50℃下冷冻干燥10h得到黑磷烯;
78.s2、将步骤s1得到的黑磷烯(50mg)超声分散在n,n-二甲基甲酰胺(dmf,10ml)中,超声功率95%,时间30分钟;然后向黑磷烯的分散液中加入聚叠氮缩水甘油醚(gap,3g),250rpm机械搅拌处理1小时,制得均匀的黑磷烯混合液,整个过程在氦气下进行。
79.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氦气,重复3次,然后以5℃/min速率升温至150℃,保温36h,冷却后,将混合物以7000rpm离心10min,然后再用n,n-二甲基甲酰胺和去离子水各洗涤3次,再将其干燥3小时即得改性黑磷烯。
80.实施例4
81.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为1mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氦气氛围下,样品底部距离超声探头2cm,超声功率为45%,以超声开2s、关4s的模式工作8h,再经过7000rpm离心10min、-50℃下冷冻干燥10h得到黑磷烯;
82.s2、将步骤s1得到的黑磷烯(50mg)超声分散在n,n-二甲基甲酰胺(dmf,10ml)中,超声功率95%,时间30分钟;然后向黑磷烯的分散液中加入聚叠氮缩水甘油醚(gap,3g),250rpm机械搅拌处理1小时,制得均匀的黑磷烯混合液,整个过程在氦气下进行。
83.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氦气,重复3次,然后以5℃/min速率升温至150℃,保温48h,冷却后,将混合物以7000rpm离心10min,然后再用n,n-二甲基甲酰胺和去离子水各洗涤3次,再将其干燥3小时即得改性黑磷烯。
84.实施例5
85.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为3mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氦气氛围下,样品底部距离超声探头2cm,超声功率为55%,以超声开2s、关4s的模式工作10h,再经过8000rpm离心10min、-30℃下冷冻干燥12h得到黑磷烯;
86.s2、将步骤s1得到的黑磷烯(40mg)超声分散在二甲基亚砜(10ml)中,超声功率55%,时间40分钟;然后向黑磷烯的分散液中加入聚3-叠氮甲基-3,甲基氧杂环丁烷(2g),500rpm机械搅拌处理2小时,制得均匀的黑磷烯混合液,整个过程在氦气下进行。
87.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氦气,重复3次,然后以7℃/min速率升温至180℃,保温30h,冷却后,将混合物以6000rpm离心20min,然后再用二甲基亚砜和去离子水各洗涤3次,再将其干燥4小时即得改性黑磷烯。
88.实施例6
89.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为4mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氦气氛围下,样品底部距离超声探头2cm,超声功率为65%,以超声开2s、关4s的模式工作12h,再经过9000rpm离心10min、-20℃下冷冻干燥8h得到黑磷烯;
90.s2、将步骤s1得到的黑磷烯(30mg)超声分散在n-甲基吡咯烷酮(10ml)中,超声功率65%,时间50分钟;然后向黑磷烯的分散液中加入聚3,3-双叠氮甲基氧杂环丁烷(2g),700rpm机械搅拌处理3小时,制得均匀的黑磷烯混合液,整个过程在氦气下进行。
91.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氦气,重复3次,然后以8℃/min速率升温至180℃,保温35h,冷却后,将混合物以8000rpm离心25min,然后再用n-甲基吡咯烷酮和去离子水各洗涤4次,再将其干燥4小时即得改性黑磷烯。
92.实施例7
93.s1、将黑磷晶体在研钵中用氩气吹扫研磨至带有金属光泽的粉末,将bp粉末转移到充满氩气的烧杯中,在烧杯中加入适量无氧去离子水,配置浓度为6mg/ml的bp-水溶液。将烧杯置于冰浴之中,放入细胞破碎仪,在氦气氛围下,样品底部距离超声探头2cm,超声功率为85%,以超声开2s、关4s的模式工作18h,再经过10000rpm离心10min、-10℃下冷冻干燥11h得到黑磷烯;
94.s2、将步骤s1得到的黑磷烯(30mg)超声分散在聚乙二醇(20ml)中,超声功率95%,时间100分钟;然后向黑磷烯的分散液中加入3,3-双叠氮甲基环氧丁烷-四氢呋喃共聚醚(1.8g),1200rpm机械搅拌处理5小时,制得均匀的黑磷烯混合液,整个过程在氦气下进行。
95.s3、将步骤s2得到的黑磷烯混合物置于史莱克管中,首先使用液氮将其冷冻,然后利用真空泵将其抽为真空状态,然后通入氦气,重复3次,然后以4℃/min速率升温至200℃,保温50h,冷却后,将混合物以9000rpm离心30min,然后再用聚乙二醇和去离子水各洗涤6次,再将其干燥8小时即得改性黑磷烯。
96.下面通过具体的应用例说明本发明实施例制备的改性黑磷烯的增强效果,通过拉伸性能测试考察该增强剂的增强效果。
97.应用例1
98.将上述实施例1制备得到的含p=n基团在空气中稳定的黑磷烯按照表1中的比例加入到gap粘合剂体系(gap,n100固化剂)中制备增强gap粘合剂体系样条,50mm
×
20mm
×
4mm用于拉伸性能测试。测试结果如下表所示:
99.表1本发明应用例增强gap粘合剂体系的效果
[0100][0101]
由表1可知,gap粘合剂体系中添加了本发明实施例1的增强剂,增强效果显著提高,且随着添加量的增加力学性能也增加。
[0102]
本说明书中各个实施例采用递进的方式描述,每个实施例重点说明的都是与其他实施例的不同之处,各个实施例之间相同相似部分互相参见即可。
[0103]
对所公开的实施例的上述说明,使本领域专业技术人员能够实现或使用本发明。对这些实施例的多种修改对本领域的专业技术人员来说将是显而易见的,本文中所定义的一般原理可以在不脱离本发明的精神或范围的情况下,在其它实施例中实现。因此,本发明将不会被限制于本文所示的这些实施例,而是要符合与本文所公开的原理和新颖特点相一致的最宽的范围。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1