具有改善的刚性/冲击平衡的异相聚丙烯的制作方法
本发明涉及具有分别有利的改善的刚性/冲击平衡的异相聚丙烯组合物。此外,本发明还涉及由本发明的聚丙烯组合物制成的制品,特别是膜制品、挤出制品、吹塑成型制品或注射成型制品。最后,本发明还涉及本发明的聚丙烯组合物在制备膜制品、挤出制品、吹塑成型制品或注射成型制品(诸如小袋和袋、管材和配件、运输包装容器以及汽车外饰和内饰的组件,如仪表板、门包层、操作台、保险杠和装饰)中的用途。
背景技术:
聚合物,如聚丙烯,越来越多地被用于不同要求的应用中。同时,对满足这些应用的要求的定制的聚合物也进行着持续的研究。由于很多聚合物的性质是直接或间接相关的,即改善特定性质只能以牺牲另一种性质为代价来完成,因此这些要求具有挑战性。可以例如通过增加组合物中均聚物的结晶度和/或相对量来改善刚性。其结果是,材料变得更脆,从而导致差的冲击性质。已知可以通过将橡胶相分散在聚合物基体中从而获得异相聚丙烯组合物来提高聚丙烯的冲击强度。
这种异相丙烯共聚物包括为丙烯均聚物或无规丙烯共聚物的基体,在基体中分散有包含丙烯共聚物橡胶(弹性体)的无定形相。因此,聚丙烯基体包含不是基体的部分的(精细)分散的夹杂物,并且所述夹杂物包含弹性体。术语夹杂物表示基体和夹杂物在异相丙烯共聚物中形成了不同的相,所述夹杂物是例如通过高分辨显微镜(如电子显微镜或扫描力显微镜或原子力显微镜)或通过动态机械热分析(dmta)可见的。此外,异相聚丙烯可以在一定程度上包含结晶聚乙烯,其是制备异相丙烯共聚物而获得的副反应产物。由于热力学原因,这种结晶聚乙烯作为无定形相的夹杂物存在。
许多不同类型的异相体系已经根据不同需求被描述。
从wo2009/129873中已知显示为降低的挥发物含量、减少雾化和降低的己烷可溶物含量的具有高纯度的异相丙烯共聚物。这些高纯度异相丙烯共聚物是在特殊的齐格勒-纳塔主催化剂的存在下采用多步聚合方法进行制备,该催化剂含有与特殊的外部给体结合的低级醇和邻苯二甲酸酯的酯交换反应产物。然而,对于不含邻苯二甲酸化合物的聚合物存在普通的市场要求。
从wo2010/049448中已知另一异相丙烯共聚物。这些高纯度的异相丙烯共聚物也是在特殊的齐格勒-纳塔主催化剂的存在下以多步聚合方法制备,该催化剂含有与特殊的外部给体结合的低级醇和邻苯二甲酸酯的酯交换反应产物。然而,在该申请中描述的产物具有受限的冲击强度。
ep1358266a1公开了具有改善的光学性能和改善的冲击强度的用于包装应用的异相聚丙烯组合物,其包含丙烯均聚物或共聚物基体相,分散的弹性体乙烯-丙烯共聚物相和作为改性剂的低密度乙烯聚合物组分。成核剂是任选的添加剂。ep1358266a1没有提及挥发物和雾化。
ep1659151a1公开了包含丙烯均聚物或共聚物基体相和包含两种在乙烯含量和特性粘度上不同的弹性体乙烯-丙烯共聚物级分的分散相的异相聚丙烯组合物。所述组合物还包含乙烯含量至少为80摩尔%的低密度乙烯共聚物。用已知α-成核剂的成核是一种选择。所述组合物适合于成型,并且具有在冲击强度和刚度之间的良好平衡、足够的流动性和良好的光学性能。ep1659151a1也没有提及挥发物和雾化。
ep1801156a1公开了包含丙烯均聚物和/或共聚物基体相、分散的弹性体乙烯-丙烯共聚物相和低密度乙烯共聚物组分的异相聚丙烯组合物。特定类型的成核剂,即聚合物成核剂被公开为是必需的。所述组合物适用于热成型和薄壁包装,并且具有良好的透明度。再次,没有提及挥发物和雾化。
尽管在异相聚丙烯组合物领域中已经进行了许多开发工作,但迄今为止,没有发现相对于与减少排放(低挥发物和低雾化)相结合的在韧性和冲击方面的良好平衡的聚合物组合物。
因此,仍然需要异相聚丙烯组合物,其在保持可接受的(低温)耐冲击性的同时具有高韧性/刚度和减少的排放,即低挥发物和低雾化。
因此,本发明的目的是提供这种材料。
技术实现要素:
本发明基于以下发现:上述目的可以通过特定的异相聚丙烯组合物实现,该组合物包括:
(a)70至90重量%的结晶全同立构丙烯均聚物基体,其具有大于96摩尔%的通过13c-nmr光谱测定的五单元组浓度和在0.5至50g/10min的范围内的根据iso1133在230℃和2.16kg负荷下测定的基体熔体流动速率(mfrm),
(b)10至30重量%的主要为无定形的丙烯共聚物,其具有32至50重量%的乙烯和/或具有4至10个碳原子的α-烯烃,作为分散的颗粒存在于组合物中,和
(c)0至5.0重量%的结晶乙烯共聚物,其具有含3至10个碳原子的α-烯烃,作为(b)的分散颗粒的夹杂物存在于组合物中,
(d)0至1.0重量%的用于全同立构聚丙烯的α-相和/或γ-相的α成核剂,
所述组合物的特征还在于0.5至45g/10min范围内的根据iso1133在230℃和2.16kg负荷下测定的总熔体流动速率(mfrt),在11至27重量%范围内的根据iso16152在25℃下测定的二甲苯可溶物级分(xcs),和≤1.0的mfrt/mfrm的比。
组合物的各个组分的百分数的总和等于100%。
与其它异相聚丙烯组合物相比,特别是组分(a)和(b)的特殊组合产生了具有改善的韧性/冲击平衡以及降低的排放(即低挥发物和低雾化)的组合物。
在本发明的第一实施方案中,异相聚丙烯组合物不含邻苯二甲酸酯类以及它们各自的分解产物,优选地,异相聚丙烯组合物不含邻苯二甲酸化合物及它们各自的分解产物。
根据本发明,术语“邻苯二甲酸化合物”是指邻苯二甲酸(cas号88-99-3)、其与脂肪族的、脂环族的和芳族的醇类的单酯类和二酯类以及邻苯二甲酸酐。
在另一方面,本发明涉及所述组合物在制备膜制品、挤出制品、吹塑成型制品或注射成型制品(诸如小袋和袋、管材和配件、运输包装容器以及汽车外饰和内饰的组件,如仪表板、门包层、操作台、保险杠和装饰)中的用途。
在另一方面,本发明涉及由本发明的聚丙烯组合物制成的制品,特别是膜制品或挤出制品、吹塑成型制品或注射成型制品。
具体实施方式
在下文中将更详细地定义各个组分。
本发明的特定异相聚丙烯组合物至少包括组分(a)和组分(b)。
组分(a):
该特定异相聚丙烯组合物的组分(a)是形成异相聚丙烯组合物的基体的结晶全同立构丙烯均聚物。
在本发明中所用的术语均聚物涉及由基本上、即至少97重量%、优选为至少98重量%、更优选为至少99重量%、还更优选为至少99.8重量%的丙烯单元组成的聚丙烯。在优选的实施方案中,在丙烯均聚物中只有丙烯单元是可检测到的。
丙烯均聚物基体是全同立构的,具有高五单元组浓度,即高于96摩尔%的五单元组浓度,如至少96.3摩尔%的五单元组浓度。五单元组浓度优选为至少96.5摩尔%至99.9%、更优选为至少96.7摩尔%至99.8%。
丙烯均聚物基体的熔体流动速率mfr2(iso1133;230℃;2.16kg)在0.5至50g/10min的范围内、优选在0.7至45g/10min的范围内、更优选在0.9至42g/10min的范围内。
基体的mfr2被称为基体熔体流动速率(mfrm)。
此外,优选的是,丙烯均聚物基体的二甲苯可溶物的量不是太高。二甲苯可溶物是在冷二甲苯中可溶的聚合物部分,其通过在沸腾的二甲苯中溶解并将不溶解的部分从冷却溶液(根据iso16152在25℃下测定)中结晶来确定。二甲苯可溶物级分包含低立体规整性的聚合物链,且表征非结晶区域的量。因此,优选的是,丙烯均聚物基体的二甲苯可溶物级分在0.5重量%至3.0重量%的范围内,更优选在0.7重量%至2.5重量%的范围内。在甚至更优选的实施方案中,二甲苯可溶物级分在0.8重量%至2.3重量%的范围内。
丙烯均聚物具有根据iso11357通过dsc分析测定的熔融温度tm1和熔融焓hm1。
优选地,丙烯均聚物的tm1在160℃至170℃的范围内,更优选在161℃至169℃的范围内,最优选在162℃至168℃的范围内。
优选地,丙烯均聚物的hm1在70至100j/g的范围内,更优选在70至95j/g的范围内,最优选在70至92j/g的范围内。
丙烯均聚物基体可以是单峰或多峰的,如双峰的。
优选地,该丙烯均聚物基体是多峰的,特别是双峰的。
关于单峰和多峰(如双峰)的定义,请参考下文的定义。
当丙烯均聚物基体包括两种以上不同的丙烯聚合物时,它们可以是具有不同单体组成和/或具有不同分子量分布的聚合物。这些组分可以具有相同或不同的单体组成和立构规整度。
当丙烯均聚物基体相在分子量分布上是单峰的时候,其可以由单阶段方法制备,例如,在淤浆或气相反应器中的淤浆或气相方法。优选地,单峰基体相作为淤浆聚合进行聚合。可选地,单峰基体可以由多阶段方法制备,在每阶段方法中采用导致类似聚合物性质的方法条件。
如果丙烯均聚物基体具有多峰或双峰特性,则可以通过将不同聚合物类型,即不同分子量和/或共聚单体含量的聚合物共混来制备丙烯均聚物基体。然而在这种情况下,优选的是,聚丙烯基体的聚合物组分是以连续步骤方法采用串联配置的反应器并在不同的反应条件下操作来制备的。因此,在特定反应器中制备的各个级分会具有其各自的分子量分布和/或共聚单体含量分布。
当将这些级分的分布曲线(分子量或共聚单体含量)叠加以获得最终聚合物的分子量分布曲线或共聚单体含量分布曲线时,这些曲线可以显示出两个以上最大值或当与各个级分的曲线进行比较时显示出至少明显变宽。根据步骤的数量,在两个或更多个连续步骤中制备的这种聚合物被称为双峰或多峰。
组分(b)
该特定异相聚丙烯组合物的组分(b)是主要为无定形丙烯共聚物,其作为分散颗粒存在于组合物中。(即分散相)
用于丙烯共聚物的合适的共聚单体是乙烯和/或具有4至10个碳原子的α-烯烃。
合适的c4-c10的α-烯烃是1-丁烯、1-戊烯、1-己烯、1-庚烯和1-辛烯。
优选地,组分(b)是丙烯和乙烯的共聚物。
组分(b)中乙烯和/或具有4-10个碳原子的α-烯烃的量在32至50重量%的范围内,优选在34至50重量%的范围内,如在36至50重量%的范围内。
与丙烯均聚物基体一样,分散相可以是单峰的或多峰的,如双峰的。
在一个实施方案中,分散相是单峰的。更具体地,鉴于特性粘度和/或共聚单体分布,该分散相优选是单峰的。关于单峰和多峰(如双峰)的定义,请参考上述定义。
组分(c)
作为组分(c)的具有3至10个碳原子的α-烯烃的结晶乙烯共聚物是任选存在的。
具有3至10个碳原子的α-烯烃例如是丙烯、1-丁烯、1-戊烯、1-己烯、1-庚烯和1-辛烯。
结晶乙烯共聚物是制备异相聚丙烯组合物而获得的副反应产物。由于热力学原因,该结晶乙烯共聚物作为夹杂物存在于无定形相中。
结晶乙烯共聚物具有根据iso11357通过dsc分析测定的熔融温度tm2和熔融焓hm2。
优选地,结晶乙烯共聚物的tm2在105℃至130℃的范围内,更优选在110℃至127℃的范围内,最优选在112℃至124℃的范围内。
优选地,结晶乙烯共聚物的hm2小于7j/g,更优选地小于6j/g,最优选地小于5j/g。
组分(d)
作为组分(d)的用于α-相和/或γ-相的全同立构聚丙烯的α成核剂是任选存在的。
众所周知的是,不同类型的晶体成核剂将不同地影响聚合物的晶体结构,增加全同立构聚丙烯的特定晶型(如单斜的α-晶型、准六方的β-晶型和正交的γ-晶型)的存在和相对量。
虽然聚合物结构将影响特定成核的表现程度,但所形成的晶体的类型将由成核剂决定。
α-成核剂(d)(如果存在)通常以0.0001至1.0重量%、优选为0.0005至0.8重量%、更优选为0.001至0.5重量%的少量加入。
α-成核剂(d)可以是聚丙烯的单斜的α-晶型和/或正交的γ-晶型的成核剂的任意化合物。
一般来说,可以区分出两类α-成核剂,即颗粒成核剂和可溶性成核剂。
颗粒成核剂显示出常规的分散机理,其中粒度和对聚合物的极性差异是决定性的。此类的实例是无机成核剂,如滑石,还有有机成核剂,如苯甲酸钠、有机磷酸盐类和对叔丁基苯甲酸的盐,以及聚合成核剂,如聚合的乙烯基化合物(诸如聚乙烯基环己烷或聚四氟乙烯)。关于这些成核剂的进一步细节可在例如wo99/24479和wo99/24501中找到。
可溶性成核剂是具有在加热时溶解并在冷却时重结晶的顺序的定义分散度的成核剂。在后一种情况下,溶解度和所得到的晶体形状对于效率是决定性的。这类的实例是如山梨醇衍生物(例如二(烷基亚苄基)山梨醇类,如1,3:2,4-25二亚苄基山梨醇、1,3:2,4二(4-甲基亚苄基)山梨醇、1,3:2,4-二(4-乙基亚苄基)山梨醇和1,3:2,4-双(3,4-二甲基亚苄基)山梨醇)和诺尼醇衍生物(例如1,2,3-三脱氧-4,6:5,7-双-o-[(4-丙基苯基)亚甲基]诺尼醇)和苯-三酰胺类(如取代的1,3,5-苯三酰胺类,如n,n',n”-三-叔丁基-1,3,5-苯三羧酰胺、n,n',n”-三-环己基-1,3,5-苯-三羧酰胺和n-[3,5-双-(2,2-二甲基-丙酰基氨基)-苯基]-2,2-二甲基-丙酰胺)的成核剂。
然而,在异相聚丙烯组合物包括α-成核剂的情况下,异相聚丙烯组合物优选地具有比非成核异相聚丙烯组合物的结晶温度高的结晶温度,由此根据iso11357通过dsc分析测定,成核的异相聚丙烯组合物的结晶温度高于120℃。
异相组合物
本发明的异相聚丙烯组合物的特征还在于在0.5至45g/10min的范围内、优选在0.6至35g/10min的范围内、更优选在0.7至30g/10min的范围内和甚至更优选在0.8至25g/10min的范围内的总熔体流动速率(mfrt)(iso1133;230℃;2.16kg)。
异相聚丙烯组合物的总熔体流动速率与丙烯均聚物基体的熔体流动速率的比值mfrt/mfrm为≤1.0。
优选地,比值mfrt/mfrm在0.3至1.0的范围内,更优选地在0.4至0.9的范围内。
异相聚丙烯组合物的根据iso16152(25℃)测定的二甲苯冷可溶物(xcs)级分在11.0至27.0重量%的范围内,优选在13.0至27.0重量%的范围内,更优选在14.0至26.0重量%的范围内。
此外,应当理解的是,异相聚丙烯组合物的二甲苯冷可溶物(xcs)级分由其特性粘度进行说明。
对于本发明,应当理解的是,异相聚丙烯组合物的二甲苯冷可溶物级分(xcs)具有在1.2至低于4.5dl/g的范围内、优选在1.5至4.0dl/g的范围内、更优选在1.7至低于3.8dl/g的范围内的根据iso1628/1(在135℃在萘烷中)测定的特性粘度(iv)。
另外,优选的是,异相聚丙烯组合物的二甲苯冷可溶物(xcs)级分的共聚单体含量、优选为乙烯含量在35.0至52.0重量%的范围内、优选在37.0至52.0重量%的范围内,且更优选在39.0至50.0重量%的范围内。
存在于二甲苯冷可溶物(xcs)级分中的共聚单体是上文中对丙烯共聚物(组分b)所定义的那些级分。在一个优选的实施方案中,共聚单体仅为乙烯。
还应当理解的是,在全部异相聚丙烯组合物中,共聚单体的总含量(即乙烯和具有4-10个碳原子的α-烯烃的含量的总和)是相当适中的。
因此,优选的是,异相聚丙烯组合物的总共聚单体含量、优选为乙烯含量在3.5至17.0重量%的范围内,优选在4.0至16.5重量%的范围内,更优选在4.5至16.2重量%的范围内。
此外,本发明的异相聚丙烯组合物具有至少第一玻璃化转变温度tg(1)和第二玻璃化转变温度tg(2),其中所述第一玻璃化转变温度tg(1)高于第二玻璃化转变温度tg(2)。玻璃化转变温度tg是根据iso6721-7通过动态机械热分析(dmta)测定的。
因此,异相聚丙烯组合物具有在-4至+4℃范围内的第一玻璃化转变温度tg(1)和在-65至-50℃范围内的第二玻璃化转变温度tg(2)。
异相聚丙烯组合物的多相结构(主要是分散在基体中的无定形丙烯共聚物)可以通过至少两个不同的玻璃化转变温度的存在来确定。较高的第一玻璃化转变温度(tg(1))表示基体,即结晶聚丙烯均聚物,而较低的第二玻璃化转变温度(tg(2))反映异相聚丙烯组合物的主要为无定形的丙烯共聚物。
优选地,第一玻璃化转变温度tg(1)在-3至+3℃的范围内,更优选在-2至+2℃的范围内。
第二玻璃化转变温度tg(2)优选在-62至-53℃的范围内,更优选在-60至-54℃的范围内。
本发明的异相聚丙烯组合物具有在800至1700mpa范围内、优选在830至1650mpa范围内、更优选在850至1600mpa范围内的在80×10×4mm3的注射成型试样上根据iso178测定的弯曲模量(fm)。
此外,本发明的异相聚丙烯组合物优选满足不等式
fm[mpa]>1660-33.4*xcs[重量%]
其中xcs是异相聚丙烯组合物的根据iso16152在25℃下测定的总二甲苯可溶物级分(xcs)。
根据iso179-1ea在23℃下测量,该异相聚丙烯组合物的简支梁缺口冲击强度在4.0至80.0kj/m2的范围内、优选在4.5至75.0kj/m2的范围内和更优选在5.0至70.0kj/m2的范围内。
根据iso179-1ea在-20℃下测量,该异相聚丙烯组合物的简支梁缺口冲击强度优选在3.0至10.0kj/m2的范围内,优选在3.5至9.0kj/m2的范围内,更优选在3.8至8.5kj/m2的范围内。
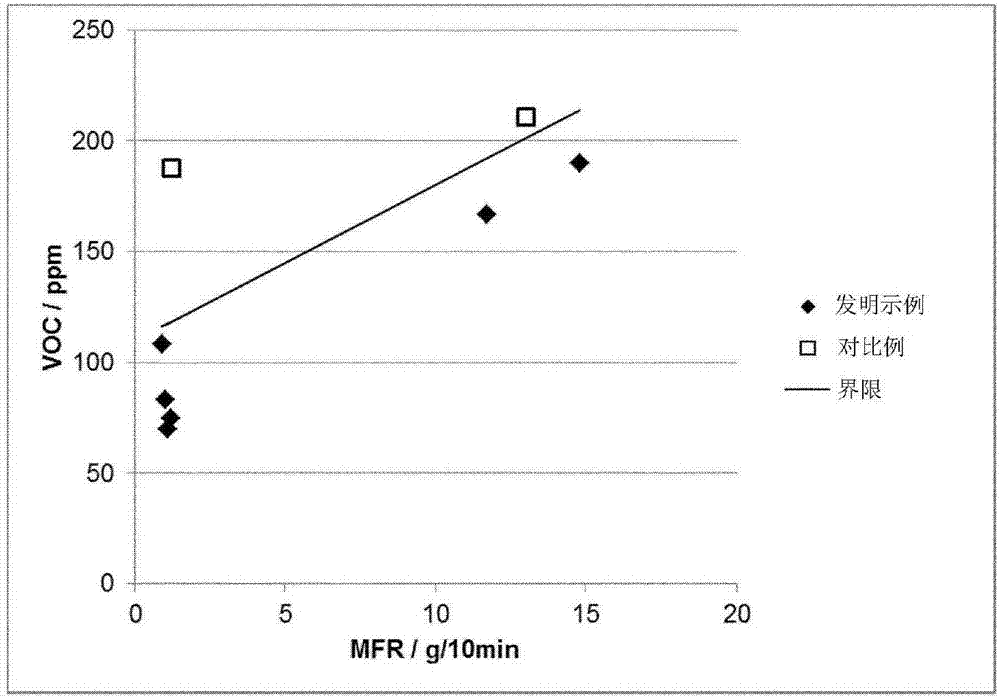
本发明的异相聚丙烯组合物具有等于或低于250ppm、优选为等于或低于220ppm、更优选为等于或低于200ppm的根据vda278:2002测量的voc值。
voc是挥发性有机化合物(voc)的量[以ppm计]。
本发明的异相聚丙烯组合物具有等于或低于300ppm,优选为等于或低于280ppm,更优选为等于或低于260ppm的根据vda278:2002测量的fog值。
fog是雾化化合物(fog)的量[以ppm计]。
此外,本发明的异相聚丙烯组合物优选地满足不等式
voc[ppm]<110+7.0*mfrt[g/10min]
其中mfrt是根据iso1133在230℃和2.16kg负荷下测定的所述组合物的总熔体流动速率。
在优选的实施方案中,该异相聚丙烯组合物优选不含邻苯二甲酸酯类以及它们各自的分解产物,即通常作为用于制备异相聚丙烯组合物的齐格勒-纳塔催化剂的内部给体的邻苯二甲酸酯类。优选地,异相聚丙烯组合物不含邻苯二甲酸化合物以及它们各自的分解产物,即通常作为齐格勒-纳塔催化剂的内部给体的邻苯二甲酸化合物。
术语“不含”邻苯二甲酸酯类,优选为邻苯二甲酸化合物,在本发明中的意义是指异相聚丙烯组合物,在其中检测不到来自齐格勒-纳塔催化剂的邻苯二甲酸酯类以及相应的分解产物,优选地检测不到邻苯二甲酸化合物以及相应的分解产物。
本发明的异相聚丙烯组合物由组分(a)和(b)和任选的组分(c)和(d)组成。
组分(a)以70至90重量%,优选73至87重量%,更优选74至86重量%的量存在。
组分(b)以30至10重量%,优选27至13重量%,更优选26至14重量%的量存在。
组分(c)以0至5.0重量%,优选0.1至4.0重量%,更优选0.2至3.0重量%的量存在。
组分(d)以0至1.0重量%,优选0至0.8重量%,更优选0至0.5重量%的量存在。
根据其它级分或添加剂的存在,级分(a)、(b)、(c)和(d)的总和为100重量%或低于100重量%。根据本发明,本文所用的重量百分比(重量%)的范围限定了各级分或组分的基于整个异相聚丙烯组合物的量。所有级分和组分一共为100重量%。
根据本发明,除了聚合物组分和α-成核剂(d)之外,该异相聚丙烯组合物可以包括其它非聚合物组分,例如用于不同目的的添加剂。
以下是任选的添加剂:工艺和热稳定剂、能够保持透明度的颜料和其它着色剂、抗氧化剂、抗静电剂、增滑剂、防粘连剂、uv稳定剂和酸清除剂。
根据添加剂的类型,可以基于异相聚丙烯组合物的重量加入0.001至2.0重量%的量的这些添加剂。
异相聚丙烯组合物的制备
该异相聚丙烯组合物可以包括至少两个串联连接的反应器的多阶段方法制备,其中首先制备聚丙烯均聚物基体(a),并且在随后的步骤中,在基体(a)的存在下制备丙烯共聚物(b)或在它们聚合之后通过将基体聚合物(a)与丙烯共聚物(b)进行共混来制备丙烯共聚物(b)。
然而,更理想地,异相聚丙烯组合物以多阶段方法制备。
在优选实施方案中,在至少一个淤浆反应器中制备聚丙烯均聚物基体(a),然后在至少一个气相反应器中制备丙烯共聚物(b)。
因此,通常可以在具有任选的第四反应器的级联的至少2个反应器至3个反应器中制备本发明的异相聚丙烯组合物,其中第一反应器是液体本体反应器,优选为环流式设计的液体本体反应器,并且所有后续反应器为气相反应器,优选为流化床设计的气相反应器。
优选地,在前两个反应器中制备的组分是可结晶的丙烯均聚物(获得基体),而在第三反应器中制备的组分是具有较高共聚单体量的主要为无定形的共聚物。任选地,可以在第四反应器中制备其它组分,其也是主要为无定形的共聚物或结晶乙烯均聚物或共聚物。
根据具体的实施方案,通过省略第二反应器或不使用第四反应器,仅使用3个反应器。
根据另一具体实施方案,仅使用第一和第三反应器。
优选的是,
(a)在第一反应器中聚合丙烯得到第一丙烯均聚物级分,
(b)将所述第一丙烯均聚物级分转移至第二反应器中,
(c)在第一丙烯均聚物级分的存在下,在所述第二反应器中聚合另外的丙烯,得到第二丙烯均聚物级分,所述第一丙烯均聚物级分和所述第二丙烯均聚物级分形成基体(a),
(d)将所述基体(a)转移至第三反应器中,
(e)在基体(a)的存在下在所述第三反应器中将丙烯与乙烯和/或c4-c10α-烯烃聚合,得到主要为无定形的丙烯共聚物(b),所述基体(a)和所述主要为无定形的丙烯共聚物(b)形成异相聚丙烯组合物。
如上文所述,通过使用串联配置并在不同条件下工作的环流式反应器和至少一个气相反应器,可以得到多峰(例如双峰)的丙烯均聚物基体(a)。
第一反应器优选地为淤浆反应器且可以是任何本体或淤浆中操作的连续或简单搅拌的间歇搅拌釜式反应器或环流式反应器。本体是指在包括至少60%(重量/重量)单体的反应介质中的聚合。根据本发明,淤浆反应器优选地是(本体)环流式反应器。
第二反应器和第三反应器优选地是气相反应器。这种气相反应器可以是任何机械混合或流化床反应器。优选地,气相反应器包括气体速度至少为0.2米/秒的机械搅拌流化床反应器。因此,可以理解的是,气相反应器是(优选地具有机械搅拌器的)流化床反应器。
因此,在一个优选的实施方案中,第一反应器是淤浆反应器,如环流式反应器,而第二反应器和第三反应器是气相反应器。因此对于本发明方法,至少三个、优选三个聚合反应器,即淤浆反应器(如环流式反应器)、第一气相反应器和第二气相反应器串联连接使用。如果需要,在淤浆反应器前放置预聚合反应器。
优选的多阶段方法为如由borealis开发(称为
另一合适的淤浆-气相方法是basell的
优选地,在本发明用于制备如上定义的异相聚丙烯共聚物的方法中,第一反应器,即淤浆反应器,如环流式反应器的条件可以如下:
-温度在50℃至110℃范围内,优选地在60℃和100℃之间,更优选在68℃和95℃之间,
-压力在20巴至80巴范围内,优选地在40巴和70巴之间,
-可以以本身已知的方法添加氢气以控制摩尔质量。
随后,将第一反应器的反应混合物转移至第二反应器,即气相反应器,其中条件优选地为如下:
-温度在50℃至130℃范围内,优选地在60℃和100℃之间,
-压力在5巴至50巴范围内,优选地在15巴和35巴之间,
-可以以本身已知的方法添加氢气以控制摩尔质量。
第三反应器中的条件与第二反应器相似。
在三个反应区中的停留时间可以不同。
在用于制备异相聚丙烯组合物的方法的一个实施方案中,在本体反应器,例如环流式反应器中的停留时间在0.1至3.5小时范围内,例如0.15至3.0小时,在气相反应器中的停留时间通常为0.2至6.0小时,如0.5至5.0小时。
如果需要,聚合可以以已知的方式在超临界条件下在第一反应器,即在淤浆反应器,如环流式反应器中进行,和/或以冷凝模式在气相反应器中进行。
优选地,该方法还包括使用如下详细描述的催化剂体系的预聚合步骤,该催化剂体系包括齐格勒-纳塔主催化剂、外部给体和任选的助催化剂。
在一个优选的实施方案中,预聚合步骤在溶解有微量其它反应物和任选的惰性组分的液态丙烯(即液相主要包括丙烯)中以本体淤浆聚合的方式进行。
预聚合反应通常在10至60℃、优选在15至50℃、更优选在20至45℃的温度下进行。
预聚合反应器中的压力不是关键的,但必须足够高以维持反应混合物为液相。因此,压力可以为20至100巴,例如30至70巴。
优选地将催化剂组分全部引入至预聚合步骤。然而,在固体催化剂组分(i)和助催化剂(ii)可以分别进料时,可以将仅部分助催化剂引入到预聚合阶段,将其余部分引入到随后的聚合阶段。并且,在这种情况下,有必要将使在预聚合阶段中获得充分的聚合反应的量的助催化剂引入到预聚合阶段中。
也可以在预聚合阶段添加其它组分。因此,如在本领域中已知的,可以在预聚合阶段添加氢气以控制预聚物的分子量。此外,可以使用抗静电添加剂来防止颗粒彼此粘附或粘附到反应器的壁上。
预聚合条件和反应参数的精确控制在本领域的技术范围内。
根据本发明,如上所述,在催化剂体系的存在下通过多阶段聚合方法得到了异相聚丙烯组合物。
如上所述,在制备如上定义的异相聚丙烯组合物的具体方法中必须使用特定的齐格勒-纳塔催化剂。
因此,现在将更详细地描述齐格勒-纳塔催化剂。
在本发明中所用的催化剂是固体齐格勒-纳塔催化剂,其包括iupac的第4至6族的过渡金属(如钛)的化合物、第2族金属(如镁)化合物和内部给体,其中内部给体优选为非邻苯二甲酸化合物,更优选为非邻苯二甲酸酯,还更优选为如下文更详细描述的非邻苯二甲酸二羧酸的二酯。因此,催化剂完全不含不需要的邻苯二甲酸化合物。此外,固体催化剂不含任何外部载体材料,如二氧化硅或mgcl2,而催化剂是自负载的。
齐格勒-纳塔催化剂(zn-c)可以通过其制备方式进一步地限定。
因此,齐格勒-纳塔催化剂(zn-c)优选地通过包括以下步骤的方法获得:
a)
a1)提供至少第2族金属烷氧基化合物(ax)的溶液,其中第2族金属烷氧基化合物(ax)是第2族金属化合物与一元醇(a)任选地在有机液体反应介质中的反应产物,其中一元醇(a)除了羟基部分外还包括至少一个醚部分;或
a2)提供至少第2族金属烷氧基化合物(ax')的溶液,其中第2族金属烷氧基化合物(ax')是第2族金属化合物(mc)与一元醇(a)和式roh的一元醇(b)的醇混合物任选地在有机液体反应介质中的反应产物;或
a3)提供第2族金属烷氧基化合物(ax)和第2族金属烷氧基化合物(bx)的混合物的溶液,其中第2族金属烷氧基化合物(bx)是第2族金属化合物与一元醇(b)任选地在有机液体反应介质中的反应产物;或
a4)提供式m(or1)n(or2)mx2-n-m的第2族金属烷氧基化合物或第2族金属烷氧基化合物m(or1)n’x2-n’和m(or2)m’x2-m’的混合物的溶液,其中m是第2族金属,x是卤素,r1和r2是c2至c16碳原子的不同烷基基团,且0<n<2,0<m<2且n+m+(2-n-m)=2,条件是n和m≠0,0<n’<2且0<m’<2;和
b)将步骤a)的所述溶液加入到第4至6族过渡金属的至少一种化合物中,和
c)获得固体催化剂组分颗粒,
和在步骤c)之前的任何步骤中加入内部给体,优选为非邻苯二甲酸的内部给体。
因此,优选地将内部给体或其前体加入到步骤a)的溶液中。
根据上面的过程,通过取决于物理条件的,尤其是步骤b)和c)中采用的温度的沉淀法或乳液(液/液两相体系)固化方法可以获得齐格勒-纳塔催化剂。
在这两种方法(沉淀或乳液固化)中,其催化剂化学是相同的。
在沉淀方法中,步骤a)的溶液与步骤b)中的至少一种过渡金属化合物进行结合,并且将整个反应混合物保持在至少50℃,更优选地保持在55℃至110℃的温度范围内,更优选保持在70℃至100℃温度范围内,以确保以固体颗粒形式存在的催化剂组分完全沉淀(步骤c)。
在乳液固化方法中,在步骤b)中,通常在诸如从-10℃至低于50℃、优选地从-5至30℃的较低的温度下将步骤a)的溶液加入到至少一种过渡金属化合物中。在乳液的搅拌期间,通常将温度保持在-10至低于40℃,优选地从-5至30℃。该乳液的分散相的液滴形成活性催化剂组合物。通过将乳液加热至70至150℃、优选为80至110℃的温度来适当地进行液滴的固化(步骤c)。
本发明中优选地采用以乳液固化方法制备的催化剂。
在一个优选的实施方案中,在步骤a)中,采用a2)或a3)的溶液,即(ax’)的溶液或(ax)和(bx)的混合物的溶液。
优选地,第2族金属是镁。
在催化剂制备方法的第一步骤,步骤a)中通过将镁化合物和如上所述的醇(类)进行反应可以原位制备烷氧基镁化合物(ax)、(ax’)和(bx),或者所述烷氧基镁化合物可以是单独制备的烷氧基镁化合物,或它们甚至可以是作为现成的烷氧基镁化合物而市售可得的以及本身在本发明的催化剂制备方法中所使用。
醇类(a)的说明性示例为二元醇类的单醚类(二醇单醚类)。优选的醇类(a)为c2至c4二醇单醚类,其中醚部分包含2至18个碳原子,优选地为4至12个碳原子。优选的示例为2-(2-乙基己氧基)乙醇、2-丁氧基乙醇、2-己氧基乙醇和1,3-丙二醇-单丁基醚、3-丁氧基-2-丙醇,特别优选的是2-(2-乙基己氧基)乙醇和1,3-丙二醇-单丁基醚、3-丁氧基-2-丙醇。
说明性一元醇类(b)具有式roh,r为直链或支链的c6-c10烷基残基。最优选的一元醇为2-乙基-1-己醇或辛醇。
优选地,分别采用和使用摩尔比bx:ax或b:a为8:1至2:1,更优选为5:1至3:1的烷氧基镁化合物(ax)和(bx)的混合物或醇(a)和(b)的混合物。
烷氧基镁化合物可以是如上所定义的醇(类)与选自二烷基镁、烷基烷氧基镁、二烷氧基镁、烷氧基卤化镁和烷基卤化镁的镁化合物的反应产物。烷基基团可以是相似的或不同的c1-c20烷基、优选为c2-c10烷基。典型的烷基-烷氧基镁化合物(当采用时)为乙基丁氧基镁、丁基戊氧基镁、辛基丁氧基镁和辛基辛氧基镁。优选地,采用二烷基镁。最优选的二烷基镁为丁基辛基镁或丁基乙基镁。
也有可能的是,除了醇(a)和醇(b)外,镁化合物还可以与式r”(oh)m的多元醇(c)反应以获得所述的烷氧基镁化合物。优选的多元醇类(如果采用)是r”为直链、环状或支链的c2至c10烃残基并且m为2至6的整数的醇类。
因此步骤a)的烷氧基镁化合物选自由二烷氧基镁、二芳氧基镁、烷氧基卤化镁、芳氧基卤化镁、烷基烷氧基镁、芳基烷氧基镁和烷基芳氧基镁组成的组。此外,可以采用二卤化镁和二烷氧基镁的混合物。
用于制备本发明催化剂所采用的溶剂可以选自具有5至20个碳原子、更优选地为5至12个碳原子的芳香族的和脂肪族的直链、支链和环状烃类或其混合物。适合的溶剂包含苯、甲苯、异丙苯、二甲苯、戊烷、己烷、庚烷、辛烷和壬烷。己烷类和戊烷类是特别优选的。
通常将镁化合物提供为如上所指出的溶剂中10至50重量%的溶液。通常市售可得的mg化合物,尤其是二烷基镁溶液是甲苯或庚烷中的20至40重量%的溶液。
用于制备烷氧基镁化合物的反应可在40℃至70℃的温度下进行。根据所采用的mg化合物和醇(类)选择最适合的温度。
第4族至第6族的过渡金属化合物优选地为钛化合物,最优选为如ticl4的卤化钛。
用于制备本发明中使用的催化剂的非邻苯二甲酸内部给体优选地选自非邻苯二甲酸羧(二)酸类的(二)酯类、1,3-二醚类、及其衍生物和混合物。尤其优选的给体为单不饱和二羧酸类的二酯类,特别是属于包含丙二酸酯类、马来酸酯类、琥珀酸酯类、柠康酸酯类、戊二酸酯类、环己烯-1,2-二羧酸酯类和苯甲酸酯类的酯类,以及其任意衍生物和/或混合物。优选的示例是例如取代的马来酸酯类和柠康酸酯类,最优选地为柠康酸酯类。
在乳液方法中,通过单一搅拌和任选加入(其它)溶剂和添加剂(诸如以本领域中已知的方式用于促进乳液的形成和/或稳定乳液的湍流最小化剂(tma)和/或乳化剂和/或乳化稳定剂(如表面活性剂))可以形成两相液-液体系。优选地,表面活性剂为丙烯酸聚合物或甲基丙烯酸聚合物。特别优选的是无支链的c12至c20的(甲基)丙烯酸酯类,诸如聚甲基丙烯酸(十六烷基)酯和聚甲基丙烯酸(十八烷基)酯以及它们的混合物。如果采用湍流最小化剂(tma),其优选地选自具有6至20个碳原子的α-烯烃单体的α-烯烃聚合物,如聚辛烯、聚壬烯、聚癸烯、聚十一烯或聚十二烯或它们的混合物。最优选的是聚癸烯。
用芳香族烃类和/或脂肪族烃类,优选地用甲苯、庚烷或戊烷可以对由沉淀或乳液固化方法获得的固体颗粒产物洗涤至少一次,优选地至少两次,最优选地至少三次。如通过蒸发或以氮气冲洗可以进一步对该催化剂进行干燥,或可以将其浆化为油状液体而无需任何干燥步骤。
最终得到的齐格勒-纳塔催化剂理想地以具有通常为5至200μm、优选地为10至100μm的平均粒径范围的颗粒形式存在。颗粒是以低孔隙率紧密的,并且具有低于20g/m2、更优选地低于10g/m2的表面积。通常,钛的量为催化剂组合物的1至6重量%,镁的量为催化剂组合物的10至20重量%以及给体的量为催化剂组合物的10至40重量%。
wo2012/007430、ep2610271、ep261027和ep2610272中公开了制备催化剂的详细描述,它们通过引用并入本文。
齐格勒-纳塔催化剂优选地与烷基铝助催化剂和任选地外部给体联合使用。
作为本发明聚合方法中的其它组分,优选地存在外部给体。合适的外部给体包含某些硅烷类、醚类、酯类、胺类、酮类、杂环化合物以及它们的共混物。特别优选的是使用硅烷。最优选的是使用下面通式的硅烷类:
raprbqsi(orc)(4-p-q)
其中ra、rb和rc表示烃基,特别是烷基基团或环烷基基团,并且其中p和q是从0到3范围内的数字并且它们的总和p+q等于或小于3。ra、rb和rc可彼此独立地进行选择并且可相同或不同。该硅烷类的具体示例为(叔丁基)2si(och3)2、(环己基)(甲基)si(och3)2、(苯基)2si(och3)2和(环戊基)2si(och3)2或具有以下通式的硅烷类:
si(och2ch3)3(nr3r4)
其中r3和r4可以相同或不同,代表具有1至12个碳原子的烃基。
r3和r4独立地选自由具有1至12个碳原子的直链脂肪烃基团、具有1至12个碳原子的支链脂肪烃基团和具有1至12个碳原子的环状脂肪烃基团组成的组。特别优选的是,r3和r4独立地选自甲基、乙基、正丙基、正丁基、辛基、癸基、异丙基、异丁基、异戊基、叔丁基、叔戊基、新戊基、环戊基、环己基、甲基环戊基和环庚基组成的组。
更优选地,r1和r2两者是相同的,还更优选地,r3和r4均是乙基基团。
尤其优选的外部给体为二环戊基二甲氧基硅烷给体(d给体)或环己基甲基二甲氧基硅烷给体(c给体)。
除了齐格勒-纳塔催化剂和任选的外部给体,还可以使用助催化剂。助催化剂优选地是周期表(iupac)的第13族化合物,例如有机铝(如铝化合物,像烷基铝、卤化铝或烷基卤化铝化合物)。因此,在一个具体实施方案中,助催化剂为如三乙基铝(teal)的三烷基铝、二烷基氯化铝或烷基二氯化铝或其混合物。在一个具体实施方案中,助催化剂为三乙基铝(teal)。
优选地,助催化剂(co)与外部给体(ed)之间的比率[co/ed]和/或助催化剂(co)与过渡金属(tm)之间的比率[co/tm]都应进行精心挑选。
因此,
(a)助催化剂(co)与外部给体(ed)的摩尔比[co/ed]必须在5至45的范围内,优选地在5至35的范围内,更优选地在5至25范围内;和任选地
(b)助催化剂(co)与钛化合物(tc)的摩尔比[co/tc]必须在大于80至500的范围内,优选地在100至350的范围内,更优选地在120至300范围内。
根据本发明的异相聚丙烯组合物优选在下列物质的存在下进行聚合:
(a)包括iupac的第4至6族的过渡金属的化合物(tc)、第2族金属化合物和内部给体的齐格勒-纳塔催化剂,其中所述内部给体是非邻苯二甲酸化合物,优选地是非邻苯二甲酸酯,且甚至更优选地是非邻苯二甲酸二羧酸的二酯;
(b)任选的助催化剂(co),和
(c)任选的外部给体(ed)。
优选的是,内部给体(id)选自任选的取代的丙二酸酯类、马来酸酯类、琥珀酸酯类、戊二酸酯类、环己烯-1,2-二羧酸酯类、苯甲酸酯类和它们的衍生物和/或混合物,优选地内部给体(id)是柠康酸酯。另外地或可选地,助催化剂(co)与外部给体(ed)的摩尔比[co/ed]为5至45。
如果根据本发明的异相聚丙烯组合物还包括组分(d),α-成核剂,则随后异相聚丙烯组合物被α成核。
从一系列反应器中最后的反应器收集异相聚丙烯组合物,将α-成核剂和任选的其它添加剂加入到异相聚丙烯组合物中。在通过复合上述定义的级分来制备异相聚丙烯组合物的情况下,可以将任何添加剂在所述复合步骤时一起加入或在所述复合步骤之后加入。
优选地,在挤出过程之前或过程中的一步复合过程中将这些添加剂混入组合物中。可选地,母料可以进行配制,其中首先将异相聚丙烯组合物仅仅与部分添加剂混合。
对于混合,可以使用常规的复合或共混装置,例如,班布里(banbury)混合机、双辊橡胶磨粉机、buss共捏合机或双螺杆挤出机。双螺杆挤出机可以同向旋转或反向旋转,优选为同向旋转。优选地,通过在足够高以软化和塑化聚合物的温度下将添加剂与聚合物材料一起共混来制备组合物。在挤出机的操作中使用的温度和压力是本领域已知的。通常,温度可以选自150至350℃的范围。用于挤出的压力优选为50至500巴。从挤出机回收的聚合物材料通常为粒料状。然后优选将这些粒料进行进一步加工,例如通过注射成型以制备本发明组合物的制品和产品。
异相聚丙烯组合物的用途
根据本发明的另一个实施方案,本发明的异相聚丙烯组合物用于制备膜制品、挤出制品、吹塑成型制品或注射成型制品(诸如小袋和袋、管材和配件、运输包装容器以及汽车外饰和内饰的组件,如仪表板、门包层、操作台、保险杠和装饰)中。
此外,本发明还涉及由本发明的聚丙烯组合物制备的制品,尤其是膜制品、挤出制品、吹塑成型制品或注射成型制品。
该制品通过适用于热塑性聚合物的任意常用转化方法进行制备,如注射成型、挤出吹塑成型、注射拉伸吹塑成型或流延膜挤出。
实验部分
a.测定方法
除非另有定义,以下术语的定义和测定方法适用于包括权利要求和以下示例的本发明的上述一般性描述。
通过nmr光谱对微结构的量化
定量核磁共振(nmr)谱用于该丙烯均聚物的全同规整度和区域-规整度的量化。
使用对1h和13c分别在400.15和100.62mhz下操作的brukeradvanceiii400核磁共振谱仪在溶液状态下记录定量13c{1h}nmr谱。在125℃下使用氮气用于所有气动装置,利用13c优化的10mm扩展温度探头记录所有光谱。
对于丙烯均聚物,将大约200mg的材料溶解于1,2-四氯乙烷-d2(tce-d2)中。为确保得到均匀的溶液,在加热块中制备初始样品后,将nmr管在旋转烘箱中进一步加热至少1小时。在插入磁体后,管在10hz下旋转。选择该设置主要是为了全同规整度分布量化所需的的高分辨率(busico,v.,cipullo,r.,prog.polym.sci.26(2001)443;busico,v;cipullo,r.,monaco,g.,vacatello,m.,segre,a.l.,macromolecules,30(1997)6251)。通过利用noe和双层waltz16解耦方案来利用标准单脉冲激励(zhou,z.,kuemmerle,r.,qiu,x.,redwine,d.,cong,r.,taha,a.,baugh,d.winniford,b.,j.mag.reson.187(2007)225;busico,v.,carbonniere,p.,cipullo,r.,pellecchia,r.,severn,j.,talarico,g.,macromol.rapidcommun.2007,28,11289)。每个光谱总共获得8192(8k)瞬态。
对定量的13c{1h}nmr光谱进行处理、积分,并且使用专有的计算机程序从积分来确定相关量化性质。
对于丙烯均聚物,所有化学位移都是以21.85ppm处的甲基全同立构的五单元组(mmmm)作为内部参考的。
能观察到对应区域缺陷(resconi,l.,cavallo,l.,fait,a.,piemontesi,f.,chem.rev.2000,100,1253;wang,w-j.,zhu,s.,macromolecules,33(2000),1157;cheng,h.n.,macromolecules17(1984),1950)或共聚单体的特征信号。
立构规整度分布是通过在23.6-19.7ppm之间的甲基区域求积分并校正与所关注的立体序列不相关的任何位点而被定量化的(busico,v.,cipullo,r.,prog.polym.sci.26(2001)443;busico,v.,cipullo,r.,monaco,g.,vacatello,m.,segre,a.l.,macromolecules30(1997)6251)。
特别地,区域缺陷和共聚单体对该立构规整度分布的定量的影响,通过从立体序列的特定积分区中减去代表性区域缺陷和共聚单体的积分来校正。
全同规整度在五单元组的水平上确定并被报告为全同立构五单元组(mmmm)序列相对于所有的五单元组序列的百分比:
[mmmm]%=100*(mmmm/所有五单元组的和)
2,1-赤式区域缺陷的存在是通过两个甲基位点在17.7和17.2ppm处的存在来表明以及通过其他特征位点确认。对应于其他类型的区域缺陷的特征信号没有被观察到(resconi,l.,cavallo,l.,fait,a.,piemontesi,f.,chem.rev.2000,100,1253)。
使用在17.7和17.2ppm处的两个特征甲基位点的平均积分来定量2,1-赤式区域缺陷的量:
p21e=(ie6+ie8)/2
对包括在本区域与主插入不相关的位点以及对从本区域排除的主插入位点进行校正基于甲基区域对1,2-主插入丙烯的量进行量化:
p12=ich3+p12e
将丙烯的总量定量为以主插入的丙烯和所有其它存在的区域缺陷之和:
p总=p12+p21e
将2,1-赤式区域缺陷的摩尔百分数相对于所有丙烯进行定量:
[21e]摩尔%=100*(p21e/p总)
通过nmr光谱对共聚单体的测定
采用定量核磁共振(nmr)光谱以定量聚合物的共聚单体含量和共聚单体序列分布。在溶液状态下采用brukeradvanceiii400nmr光谱仪对于1h和13c分别在400.15mhz和100.62mhz下操作来记录定量13c{1h}nmr光谱。在125℃下使用氮气用于所有气动装置,利用13c优化的10mm扩展温度探头记录所有光谱。将约200mg的材料与乙酰丙酮铬(ⅲ)(cr(acac)3)一同溶解于3ml的1,2-四氯乙烷-d2(tce-d2)中,形成65mm弛豫试剂在溶剂中的溶液(singh,g.,kothari,a.,gupta,v.,polymertesting285(2009),475)。为确保均相溶液,在加热块中制备初始样品后,将nmr管在旋转烘箱中进一步加热至少1小时。插入磁体后,管在10hz下旋转。选择该设置主要是为了高分辨率和准确的乙烯含量定量的量化需要。通过采用优化的顶锥角(tipangle)、1s的循环延迟和双层waltz16解耦方案来利用标准单脉冲激发而不采用noe(zhou,z.,kuemmerle,r.,qiu,x.,redwine,d.,cong,r.,taha,a.,baugh,d.winniford,b.,j.mag.reson.187(2007)225;busico,v.,carbonniere,p.,cipullo,r.,pellecchia,r.,severn,j.,talarico,g.,macromol.rapidcommun.2007,28,1128)。每个光谱共获得6144(6k)个瞬态。
对定量13c{1h}nmr谱进行处理、积分并采用专有的计算机程序由积分确定相关的定量性质。利用溶剂的化学位移,所有化学位移间接地参考乙烯嵌段(eee)在30.00ppm处的中心亚甲基。即使不存在这种结构单元,这种方法也允许可比较的参考。可观察到对应于乙烯的掺入的特征信号(cheng,h.n.,macromolecules17(1984),1950)。
在观察到对应于2,1-赤式区域缺陷的特征信号的情况下(如在l.resconi,l.cavallo,a.fait,f.piemontesi,chem.rev.2000,100(4),1253,cheng,h.n.,macromolecules1984,17,1950,和w-j.wang和s.zhu,macromolecules2000,331157中所描述的),区域缺陷对确定性质的影响的校正被需求。对应于其他类型的区域缺陷的特征信号未被观察到。
在13c{1h}光谱中,通过穿过整个光谱区的多个信号的积分,使用wang等人(wang,w-j.,zhu,s.,macromolecules33(2000),1157)中的方法量化共聚单体分数。该方法因其稳健的性质和在需要时说明区域缺陷的存在的能力被选择。小幅调整积分区以提高穿过遇到的共聚单体含量的整个范围的适用性。
对于在ppepp序列只能观察到孤立乙烯的体系,wang等人的方法被修改,以减少已知不存在的那些位点的非零积分的影响。这种方法减少了对这种体系的乙烯含量的过高估计,并通过减少用于确定绝对乙烯含量的位点的数目来实现:
e=0.5(sββ+sβγ+sβδ+0.5(sαβ+sαγ))
通过使用这种设置的位点,相应的积分等式变为:
e=0.5(ih+ig+0.5(ic+id))
采用wang等人的文章中所采用的相同符号(wang,w-j.,zhu,s.,macromolecules33(2000),1157)。不修改用于绝对丙烯含量的等式。
由摩尔分数计算引入的共聚单体的摩尔百分数:
e[摩尔%]=100*fe
由摩尔分数计算引入的共聚单体的重量百分数:
e[重量%]=100*(fe*28.06)/((fe*28.06)+((1-fe)*42.08))
采用kakugo等人的分析方法(kakugo,m.,naito,y.,mizunuma,k.,miyatake,t.macromolecules15(1982)1150)测定处于三单元组水平的共聚单体序列分布。选择该方法是由于其稳健的性质和稍微调整积分区域以增加更宽范围的共聚单体含量的适用性。
在室温下的二甲苯可溶物级分(xcs,重量%):可溶于二甲苯的聚合物的量在25℃下根据iso16152;第5版;2005-07-01来确定。
特性粘度(iv)
特性粘度(v)值随着聚合物分子量而增加。例如xcs的iv值是根据iso1628/1在135℃下在萘烷中测量的。
dsc分析、熔融温度(tm)、熔融焓(hm)、结晶温度(tc)和结晶焓(hc):用tainstrumentq200差示扫描量热法(dsc)在5至7mg的样品上进行测量。dsc根据iso11357/第3部分/方法c2在加热/冷却/加热循环下以10℃/min的扫描速度在-30至+225℃温度范围内运行。分别从冷却步骤中确定结晶温度(tc)和结晶焓(hc),从第二加热步骤中或在网的情况下从第一加热步骤中确定熔融温度(tm)和熔融焓(hm)。
玻璃化转变温度tg通过根据iso6721-7的动态力学热分析测定。该测量是在扭转模式下在-100℃和+150℃之间对压模样品(40×10×1mm3)以2℃/min的加热速率和1hz的频率进行。
mfr2(230℃)是根据iso1133(230℃,2.16kg负荷)来测定的。
根据iso113315(230℃,2.16kg负荷)对聚丙烯测定熔体流动速率,根据iso1133(190℃,2.16kg负荷)对聚乙烯测定熔体流动速率,并以g/10min表示。mfr表征聚合物流动性,由此表征聚合物的加工性。熔体流动速率越高,聚合物的粘度越低。
采用所测定的级分(a)和制备级分(b)后所得的混合物(“最终”)的mfr2值来计算在级分(a)存在下制备的级分(b)的mfr2:
简支梁缺口冲击强度
简支梁缺口冲击强度是根据iso1791ea在+23℃和-20℃下采用根据iso1873注射成型的测试试样(80×10×4mm)测定的。
弯曲模量:弯曲模量根据iso178使用符合eniso1873-2在23℃下注射成型的80×10×4mm3测试条上以三点弯曲进行测量。
voc/fog排放
根据vda278:2002对颗粒化合物测量voc/fog排放。挥发性有机化合物以每克样品的甲苯当量(μgte/g)进行测量。雾化以每克样品的十六烷当量(μghd/g)进行测量。
使用氦气5.0作为载气和长度为50m、直径为0.32mm和5%的苯基-甲基-硅氧烷涂层为0.52μm的柱hpultra2,以由gerstel提供的tdsa进行测量。
voc分析根据标准中列出的设备设置1进行,采用以下主要参数:流量模式不分流、最终温度为90℃;最终时间30分钟、速率60k/min。以12k/秒的加热速率和5分钟的最终时间在-150℃至+280℃的温度范围内,以1:30的流动模式分裂值对冷却井进行清洗。
使用以下gc设置用于分析:40℃等温2min,以3k/min加热至92℃,然后以5k/min加热至160℃,然后以10k/min加热至280℃,10分钟等温;流量1,3ml/min。
voc量表示c10至c15的种类。
fog分析根据标准中列出的设备设置1进行,采用以下主要参数:流量模式不分流、速率60k/min;最终温度为120℃;最终时间60分钟。以12k/秒的加热速率在-150℃至+280℃的温度范围内,以1:30的流动模式分裂值对冷却井进行清洗。使用以下gc设置用于分析:在50℃等温2min,以25k/分钟加热至160℃,然后以10k/分钟加至280℃,30分钟等温;流量1,3ml/min。
fog量表示c16至c30的种类。
b.示例
用于发明示例(ie1至ie7)的异相聚丙烯组合物的聚合方法中的催化剂的制备如下:
所用的化学品:
20%的丁基乙基镁(mg(bu)(et),bem)的甲苯溶液,由chemtura提供
2-乙基己醇,由amphochem提供
3-丁氧基-2-丙醇-(dowanoltmpnb),由dow提供
柠康酸二(2-乙基己基)酯,由synphabase提供
ticl4,由milleniumchemicals提供
甲苯,由aspokem提供
庚烷,由chevron提供
烷氧基镁化合物的制备
在20升不锈钢反应器中,在搅拌(70rpm)下,烷氧基镁溶液通过向11kg的20重量%的丁基乙基镁(mg(bu)(et))的甲苯溶液中加入4.7kg2-乙基己醇和1.2kg丁氧基丙醇的混合物而制备。在加入过程中,将反应器内容物保持在低于45℃。加入完成后,将反应混合物在60℃下继续混合(70rpm)30分钟。冷却至室温后,将2.3kg的给体柠康酸二(2-乙基己基)酯加入至温度保持在低于25℃的烷氧基镁溶液中。在搅拌(70rpm)下继续混合15分钟。
固体催化剂组分的制备
将20.3kgticl4和1.1kg甲苯加入到20升不锈钢反应器中。在350rpm下混合并将温度保持在0℃,在1.5小时内加入14.5kg示例1中制备的烷氧基镁化合物。加入1.7升的
将由此得到的催化剂与作为助催化剂的三乙基铝(teal)和作为给体的二(环戊基)二甲氧基硅烷(d-给体)一起使用。
助催化剂(co)与外部给体(ed)的摩尔比[co/ed]和助催化剂(co)与钛化合物(tc)的摩尔比[co/tc]示于表1中。
在borstar中试装置中进行聚合,该装置包括预聚合反应器、环流式反应器和两个气相反应器。聚合条件也示于表1中。
表1:发明示例的聚合
对于对比例ce1至ce3,下面的异相聚丙烯聚合物如下所述进行制备:
催化剂的制备
首先,在大气压下,0.1摩尔的mgcl2×3etoh在惰性条件下悬浮于反应器中的250ml的癸烷中。将溶液冷却至-15℃的温度并加入300ml的冷ticl4,同时保持温度处于所述水平。然后,将浆料的温度缓慢升高至20℃。在此温度下,向浆料中加入0.02摩尔的邻苯二甲酸二辛酯(dop)。在加入邻苯二甲酸酯之后,90分钟内将温度升高至135℃,将浆料静置60分钟。然后,加入另外300ml的ticl4并将温度在135℃下保持120分钟。此后,催化剂从液体中过滤,并在80℃下用300ml庚烷洗涤六次。然后,将固体催化剂组分过滤并干燥。
在诸如专利文献ep491566、ep591224和ep586390中大体描述了催化剂及其制备概念。
二(环戊基)二甲氧基硅烷(给体d)用作外部给体。ce1和ce2通过省略第一gpr,即仅在一个环流式和一个gpr中进行制备。
表2:对比例的聚合
从各个反应器中得到的产物的性质自然不是在均质材料上而是在反应器样品(局部样品)上测量的。最终树脂的性质是在均质的材料上测量,由此制备的粒料的mfr2在如下所述的挤出混合过程中测量。
所有树脂在双螺杆挤出机中使用以下添加剂进行混合:由basfag提供的0.1重量%的四(3-(3',5'-二叔丁基-4-羟基苯基)-丙酸季戊四醇酯(cas号6683-19-8,商品名irganox1010),由basfag提供的0.1重量%的磷酸三(2,4-二叔丁基苯基)酯(cas号31570-04-4,商品10名irgafos168)和由crodapolymeradditives提供的0.05重量%的硬脂酸钙(cas号1592-23-0)。
示例7的异相聚丙烯通过分别加入滑石(luzenac的steamict1ca,截留粒径(d95)为6.2μm)(示例7-1)和2,2’-亚甲基双-(4,6-二叔丁基苯基)磷酸钠(示例7-2)(na11;由basf提供的irgastabna11uh,cas号85209-91-2)进一步成核。
上述配混步骤中加入的成核剂为1.0重量%的滑石和0.2重量%的na11。
聚合物的性质被列入表3和表4中:
表3:发明示例(示例1至示例7)的聚合物性质
fm弯曲模量
n.m.未测量
表4:对比例ce1至ce3的聚合物性质
从表3和表4中可以清楚地看出,与对比例相比,本发明的异相聚丙烯组合物具有改善的刚性/冲击平衡。
从图1和图2中可进一步观察到,对比例不符合与不等式fm>1660-33.4*xcs和voc<110+7.0*mfr相关的要求。
- 还没有人留言评论。精彩留言会获得点赞!