一种胶原蛋白改性方法及使用所述方法制得的改性胶原蛋白与流程
本发明涉及生物医学材料领域,具体地涉及一种胶原蛋白改性方法及使用所述方法制得的改性胶原蛋白。
背景技术:
胶原蛋白作为一种天然的结构蛋白,自身具有优良的化学、生物特性,在生物、医学等诸多领域有着广泛的应用。但是,胶原蛋白在植入到人体之后性质不稳定,因而容易被机体所吸收降解。
为了充分利用胶原蛋白的优异性能,往往需要对胶原蛋白进行一定的改性处理以增强其稳定性,从而使其在人体内能够存在一定时间之后再降解。具体地,以胶原蛋白制备的硬脑膜补片为例,临床上需要其能在植入前期替代硬脑膜实现屏障功能,中期具有组织引导再生作用以引导自身硬脑膜形成,后期在自体硬脑膜形成过程中逐渐降解,这也就要求所使用的胶原蛋白必须具备一定程度的稳定性。
目前,主要通过交联改性胶原蛋白以提高其交联度来改善胶原蛋白的稳定性。交联改性方式主要分为物理交联和化学交联。其中,传统的物理交联主要采用真空高温来进行,这种交联方式无法达到较为理想的交联度,交联所得产品在人体内依然无法控制适合的存在时间;且热交联需要用到高温,这容易导致胶原蛋白发生变性,不利于产品的质量控制。传统的化学交联主要采用以戊二醛、甲醛、碳二亚胺等为代表的交联剂对胶原蛋白进行交联,这会造成交联剂的残留。由于交联剂在人体内也属于活性物质,当其残留大于一定剂量时容易对人体产生毒副作用。此外,相关法规也规定,对于交联剂必须进行出厂限度检查以保证产品中存在的交联剂能够被最大限度地去除,而这无疑增加了企业的管理成本。
综上所述,本领域急需一种可有效改善胶原蛋白交联度且不会引入有害残留物质的改性方法,以及一种交联度可控、稳定性优异且不含有害物质的胶原蛋白。
技术实现要素:
本发明的一目的在于提供一种可有效改善胶原蛋白交联度、基本不会引起所述胶原蛋白变性、且不会引入有害残留物质的改性方法。
本发明的另一目的在于提供一种交联度可控、稳定性优异且不含有害物质的胶原蛋白。
本发明的第一方面,提供了一种胶原蛋白改性方法,所述方法包括如下步骤:
1)提供第一溶液和交联促进剂,其中,
所述第一溶液包含胶原蛋白;
2)混合所述第一溶液和所述交联促进剂,得到第一混合物;
3)辐照处理所述第一混合物,得到经辐照处理的混合物;和
4)任选地冷冻干燥前述步骤所得产物,得到改性胶原蛋白。
在另一优选例中,所述胶原蛋白选自下组:I型胶原蛋白、Ⅲ型胶原蛋白、或其组合。
在另一优选例中,所述交联促进剂为富羟基交联促进剂。
在另一优选例中,所述富羟基交联促进剂为取代或未取代的C1-C8醇,较佳地为取代或未取代的C2-C6醇,更佳地为取代或未取代的C2-C5醇。
在另一优选例中,所述取代表示基团上的一个或多个氢原子被选自下组的取代基取代:C1-C6烷基、卤素、羟基、氨基、羧基。
在另一优选例中,当所述交联促进剂为固体时,在步骤1)之前还包括如下步骤:将所述固体交联促进剂水浴融化。
在另一优选例中,所述富羟基交联促进剂选自下组:聚乙二醇、丙三醇、乙二醇甲醚、乙二醇、或其组合。
在另一优选例中,所述聚乙二醇的分子量为200-4000,较佳地400-3000。
在另一优选例中,在步骤2)中,在所述第一混合物中,所述胶原蛋白和所述交联促进剂中羟基的摩尔比约为1-10:50000-500000。
在另一优选例中,在步骤2)中,在所述第一混合物中,所述胶原蛋白和所述交联促进剂的质量比为1-5:1-30,较佳地为1-3:3-20。
在另一优选例中,在步骤2)中,在所述第一混合物中,按重量计,所述交联促进剂的含量为2-30wt%,较佳地3-25wt%,更佳地5-15wt%。
在另一优选例中,在步骤2)中,在所述第一混合物中,按重量计,所述胶原蛋白的含量为0.1-5wt%,较佳地0.2-3wt%。
在另一优选例中,步骤3)所述辐照处理的辐照剂量为5-30kGy。
在另一优选例中,步骤3)所述辐照处理的辐照剂量为8-25kGy,较佳地10-20kGy,更佳地15-20kGy(满足交联的同时达到灭菌剂量)。
在另一优选例中,步骤3)所述辐照处理采用钴-60作为辐照靶剂量。
在另一优选例中,步骤4)所述冷冻干燥分两步进行:
1)预冻处理;和
2)干燥处理。
在另一优选例中,所述预冻处理的处理温度为0至-40℃;和/或
所述预冻处理的处理时间为0.5-5小时。
在另一优选例中,所述干燥处理的处理温度为-30至40℃;和/或
所述干燥处理的处理时间为3-20小时。
本发明的第二方面,提供了一种改性胶原蛋白,1mg所述改性胶原蛋白的吸光度≤0.25。
在另一优选例中,1mg所述改性胶原蛋白的吸光度≤0.23,较佳地≤0.22。
在另一优选例中,所述改性胶原蛋白的变性率≤5%,较佳地≤2%,更佳地≤0.5%。
在另一优选例中,所述改性胶原蛋白中交联剂的含量≤2wt%,较佳地≤0.5wt%,更佳地约为零。
在另一优选例中,所述交联剂选自下组:戊二醛、甲醛、碳二亚胺或其组合。
在另一优选例中,所述改性胶原蛋白中交联促进剂的残留量≤2wt%,较佳地≤1wt%。
在另一优选例中,所述改性胶原蛋白中的所述交联促进剂可通过水洗去除。
在另一优选例中,所述改性胶原蛋白是采用本发明第一方面所述的方法制备的。
在另一优选例中,所述改性胶原蛋白具有选自下组的一个或多个特征:
1)0.1g所述改性胶原蛋白中微生物含量≤1CFU;
2)1g所述改性胶原蛋白的吸水量≥40g;
3)所述改性胶原蛋白的拉伸强度≥1.8MPa;
4)1g所述改性胶原蛋白的体外降解时间≥10h;
5)所述改性胶原蛋白的细胞毒性<2级。
在另一优选例中,所述改性胶原蛋白具有选自下组的一个或多个特征:
1)0.1g所述改性胶原蛋白中微生物含量≤1CFU;
2)所述改性胶原蛋白的拉伸强度≥1.9MPa;
3)1g所述改性胶原蛋白的体外降解时间≥15h。
本发明的第三方面,提供了一种制品,所述制品包含本发明第二方面所述的改性胶原蛋白或由本发明第二方面所述的改性胶原蛋白组成。
在另一优选例中,所述制品选自下组:生物医学材料、心脏瓣膜膜材。
在另一优选例中,所述生物医学材料选自下组:皮肤修复胶原海绵、硬脑膜胶原补片、牙填充材料、止血材料、骨修复材料。
本发明的第四方面,提供了一种本发明第二方面所述的改性胶原蛋白的用途,用于制备生物医学材料。
应理解,在本发明范围内中,本发明的上述各技术特征和在下文(如实施例)中具体描述的各技术特征之间都可以互相组合,从而构成新的或优选的技术方案。限于篇幅,在此不再一一累述。
附图说明
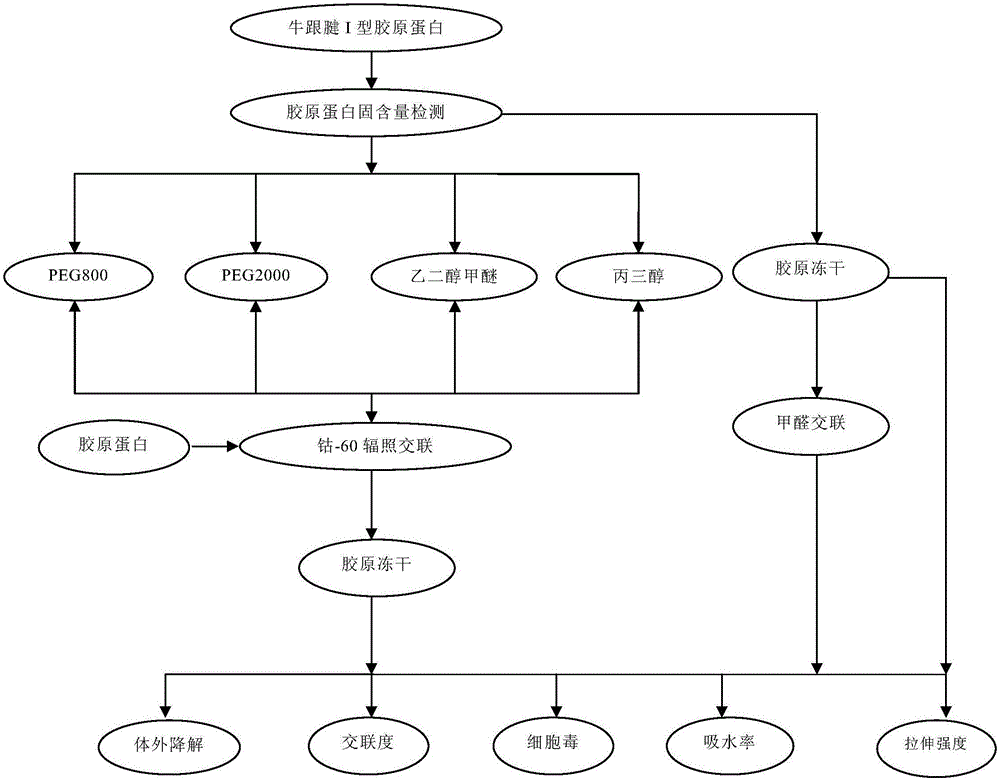
图1是本发明辐照交联和化学交联(具体为甲醛交联)的对比技术路线图。。
具体实施方式
本发明人经过长期而深入的研究,通过在待辐照处理胶原蛋白原液中添加特定比例的特定种类的交联促进剂,可在较低的辐照剂量下获得具有较高交联度的胶原蛋白。具体地,本发明人采用具有良好生物相容性的富羟基交联促进剂,在基本不引起胶原蛋白变性和/或降解且不引入有害物质的情况下,采用辐照交联技术可同时实现对胶原蛋白的改性交联和灭菌处理。在此基础上,发明人完成了本发明。
术语
如本文所用,术语“富羟基交联促进剂”、“交联促进剂”或者“促进剂”可互换使用,均指按该促进剂的质量计,羟基/分子中碳原子的含量≥70%(较佳地80%)的物质。
改性方法
本发明提供了一种胶原蛋白改性方法,所述方法包括如下步骤:
1)提供第一溶液和交联促进剂,其中,
所述第一溶液包含胶原蛋白;
2)混合所述第一溶液和所述交联促进剂,得到第一混合物;
3)辐照处理所述第一混合物,得到经辐照处理的混合物;和
4)任选地冷冻干燥前述步骤所得产物,得到改性胶原蛋白。
在另一优选例中,所述胶原蛋白包括(但并不限于):I型胶原蛋白、Ⅲ型胶原蛋白、或其组合。
在本发明中,所述交联促进剂为富羟基交联促进剂。
在另一优选例中,所述富羟基交联促进剂为取代或未取代的C1-C8醇,较佳地为取代或未取代的C2-C6醇,更佳地为取代或未取代的C2-C5醇。
在另一优选例中,所述取代表示基团上的一个或多个氢原子被包括(但并不限于)以下的取代基取代:C1-C6烷基、卤素、羟基、氨基、羧基。
在另一优选例中,当所述交联促进剂为固体时,在步骤1)之前还包括如下步骤:将所述固体交联促进剂水浴融化。
在本发明中,所述富羟基交联促进剂包括(但并不限于):聚乙二醇、丙三醇、乙二醇甲醚、乙二醇、或其组合。
在另一优选例中,所述聚乙二醇的分子量为200-4000,较佳地400-3000。
在本发明中,在步骤2)中,在所述第一混合物中,所述胶原蛋白和所述交联促进剂中羟基的摩尔比约为1-10:50000-500000。
在另一优选例中,在步骤2)中,在所述第一混合物中,所述胶原蛋白和所述交联促进剂的质量比为1-5:1-30,较佳地为1-3:3-20。
在另一优选例中,在步骤2)中,在所述第一混合物中,按重量计,所述交联促进剂的含量为2-30wt%,较佳地3-25wt%,更佳地5-15wt%。
应理解,在本发明中,在所述第一混合物中,当交联促进剂的含量高于30wt%之后,因为交联剂比例过高容易导致交联剂之间发生自交联,且容易导致其生物相容性降低,因此不利于材料的应用;当交联促进剂比例过低之时(如≤2wt%),交联剂的量无法充分对样品中存在的氨基酸残基进行交联而影响材料的性质。
在另一优选例中,在步骤2)中,在所述第一混合物中,按重量计,所述胶原蛋白的含量为0.1-5wt%,较佳地0.2-3wt%。
在本发明中,步骤3)所述辐照处理的辐照剂量为5-30kGy。
在另一优选例中,步骤3)所述辐照处理的辐照剂量为8-25kGy,较佳地10-20kGy,更佳地15-20kGy。
在另一优选例中,步骤3)所述辐照处理采用钴-60作为辐照靶剂量。
在本发明中,步骤4)所述冷冻干燥分两步进行:
1)预冻处理;和
2)干燥处理。
在另一优选例中,所述预冻处理的处理温度为0至-40℃;和/或
所述预冻处理的处理时间为0.5-5小时。
在另一优选例中,所述干燥处理的处理温度为-30至40℃;和/或
所述干燥处理的处理时间为3-20小时。
典型地,一种典型的改性方法如下所述:
1)在从牛跟腱提取的I型胶原蛋白中添加适宜种类和用量的交联促进剂;
2)通过适当辐照强度的钴-60射线辐照所得混合物以对凝胶状态的胶原蛋白进行交联处理;
3)对上述步骤所得产物在适当的温度下进行冷冻干燥,得到经改性的胶原蛋白。
本发明所述方法在不使用任何对人体具有毒副作用的交联剂的情况下,仅仅采用生物相容性良好的富羟基交联促进剂结合辐照处理就可以高效地提高胶原蛋白的交联度,进而改善胶原蛋白的稳定性,使得其降解时间显著延长。
此外,本发明所述方法在有效提高胶原蛋白交联度的同时,所述辐照作用可同时有效杀死微生物,从而避免微生物以胶原蛋白为原料增殖,这无疑降低了产品被污染的可能性。
经改性的胶原蛋白
本发明还提供了一种改性胶原蛋白,1mg所述改性胶原蛋白的吸光度≤0.25。
在另一优选例中,1mg所述改性胶原蛋白的吸光度≤0.23,较佳地≤0.22。
在另一优选例中,所述改性胶原蛋白的变性率≤5%,较佳地≤2%,更佳地≤0.5%。
在另一优选例中,所述改性胶原蛋白中交联剂的含量≤2wt%,较佳地≤0.5wt%,更佳地约为零。
在另一优选例中,所述交联剂包括(但并不限于):戊二醛、甲醛、碳二亚胺或其组合。
在另一优选例中,所述改性胶原蛋白中交联促进剂的残留量≤2wt%,较佳地≤1wt%。
在另一优选例中,所述改性胶原蛋白中的所述交联促进剂可通过水洗去除。
在另一优选例中,所述改性胶原蛋白是采用本发明所述的方法制备的。
在本发明中,所述改性胶原蛋白具有选自下组的一个或多个特征:
1)0.1g所述改性胶原蛋白中微生物含量≤1CFU;
2)1g所述改性胶原蛋白的吸水量≥40g;
3)所述改性胶原蛋白的拉伸强度≥1.8MPa;
4)1g所述改性胶原蛋白的体外降解时间≥10h;
5)所述改性胶原蛋白的细胞毒性<2级。
在另一优选例中,所述改性胶原蛋白具有选自下组的一个或多个特征:
1)0.1g所述改性胶原蛋白中微生物含量≤1CFU;
2)所述改性胶原蛋白的拉伸强度≥1.9MPa;
3)1g所述改性胶原蛋白的体外降解时间≥15h,较佳地≥50h,更佳地≥100h,更佳地≥150h,更佳地≥200h,最佳地≥210h。
应用
本发明还提供了一种制品,所述制品包含所述的改性胶原蛋白或由所述的改性胶原蛋白组成。
在另一优选例中,所述制品包括(但并不限于):生物医学材料、心脏瓣膜膜材。
在另一优选例中,所述生物医学材料包括(但并不限于):皮肤修复胶原海绵、硬脑膜胶原补片、牙填充材料、止血材料、骨修复材料。
本发明还提供了一种所述的改性胶原蛋白的用途,用于制备生物医学材料。
与现有技术相比,本发明具有以下主要优点:
(1)所述方法操作简单、安全无毒;
(2)所述方法可可控地提高胶原蛋白的交联度,进而显著延长其降解所需时间,胶原蛋白的稳定性得到显著提升;
(3)在交联促进剂的作用下,所述方法可在较低的辐照剂量下获得具有较高交联度的胶原蛋白;
(4)所述方法可一步实现交联和灭菌作用;
(5)所述方法可有效提升胶原蛋白的吸水率、拉伸强度、顶破强度等物理性能;
(6)所述方法不会在胶原蛋白中引入对人体有害的物质;
(7)所述经改性的胶原蛋白具有高交联度、高吸水率、高拉升强度和长降解时间;
(8)所述经改性的胶原蛋白基本不含对人体有害的物质,故其细胞毒性符合人体生物材料植入的要求;
(9)相比于其它物理交联方法,本发明所述交联方式能更好地提升样品的物理性能;相比其它化学交联方法,本发明所述交联方式可显著降低产品的检验成本且达到不亚于其它化学交联的效果。
下面结合具体实施例,进一步阐述本发明。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,通常按照常规条件或按照制造厂商所建议的条件。除非另外说明,否则百分比和份数按重量计算。
除非另行定义,文中所使用的所有专业与科学用语与本领域熟练人员所熟悉的意义相同。此外,任何与所记载内容相似或均等的方法及材料皆可应用于本发明方法中。文中所述的较佳实施方法与材料仅作示范之用。
实验原料
胶原蛋白原液:从牛跟腱中采用胃蛋白酶在醋酸环境下提取。所提取的胶原蛋白用纯化水进行稀释,采用固含量进行检测,配制成为0.5%~2.4wt%的胶原原液。
PEG800:采用市售的分析纯PEG800试剂,其在常温下为液体。
PEG2000:采用市售的分析纯PEG2000试剂,其在常温下为固体,使用前在沸水浴中预热融化成为液体,用纯化水进行配置。
乙二醇甲醚:采用市售分析纯试剂,无色、具醚气味液体,溶于纯化水中。
丙三醇:采用市售分析纯试剂,无色,溶于纯化水中。
实验条件
辐照靶:钴-60
辐射剂量:1-30kGy
辐射处理时间:30-100min
通用测试方法
微生物含量
(1)微生物限度检测:采用2015版本药典四部通则1105非无菌产品微生物限度检查:微生物计数法的方法对样品中的初始污染菌进行计数,采用薄膜过滤法,方法如下:所采用的滤膜孔径为0.22μm,直径一般为50mm,滤器及滤膜使用前采用高压蒸汽灭菌。使用时,应保证滤膜在过滤前后的完整性。将样品用剪刀剪碎,用生理盐水蛋白胨洗脱液对样品中的微生物进行浸提,然后用过滤装置对供试液过滤,每张滤膜每次冲洗量为100ml。过滤完成后,转移滤膜菌面朝上贴于胰酪大豆胨琼脂培养基平板上于30至35℃进行培养。培养三天后对菌落计数。
(2)无菌检验:对辐照交联并二次辐照后进行冻干的样品进行无菌检验,无菌检验的方法采用2015版本药典四部通则1101无菌检査法。采用直接接种法,方法如下:取规定量供试品分别等量接种至硫乙醇酸盐流体培养基和胰酪大豆胨液体培养基中,每个容器中培养基的用量应符合接种的供试品体积不得大于培养基体积的10%,同时,硫乙醇酸盐流体培养基每管装量不少于15ml,胰酪大豆胨液体培养基每管装量不少于10ml。供试品检査时,培养基的用量和髙度同方法适用性试验。将上述接种供试品后的培养基容器分别按各培养基规定的温度培养14天;培养期间应逐日观察并记录是否有菌生长。如在加入供试品后或在培养过程中,培养基出现浑浊,培养14天后,不能从外观上判断有无微生物生长,可取该培养液适量转种至同种新鲜培养基中,培养3天,观察接种的同种新鲜培养基是否再出现浑浊;或取培养液涂片,染色,镜检,判断是否有菌。
吸水量
吸水量检测用以表征样品对于液体的吸收能力,在生物医学材料(如硬脑膜补片)中,要求其必须能够吸收渗漏的脑脊液等。
具体检测方法如下:
将样品用电子天平精密称重(记为W0)后放入生理盐水中,待样品被生理盐水充分地饱和后,用镊子将样品拿起,直到不继续滴水为止,称量吸水后样品的总重(记为W1)。
吸水量记为(W1-W0)/W0,用以表征每克样品的吸水性能。
拉伸强度
将厚度基本相同(胶原蛋白浓度相同时)的样品裁剪成20mm宽和80mm长的长条状后,将其放置在温度23℃、湿度50%的环境下恒定24小时。设定拉伸试验仪(仪器型号:设备传感器力学限度为50kg)夹持距离为50mm±1mm,在夹持钳中心位置夹持样品以测定样品拉伸强度。
体外降解
配制生理盐水,用生理盐水配制浓度为20cdu/ml的胶原蛋白酶,采用0.22μm的微孔滤膜过滤灭菌。采用无菌技术将样品剪成1.0cm×2.0cm的长方形,用电子天平称量样品,并置于相应无菌15mlEP管中,每个样品三个重复。按照样品质量:胶原蛋白酶溶液体积=1:150(w/v)的比例加入胶原蛋白酶溶液。将试管置于37℃恒温震荡水浴锅中,100rpm震荡。开始酶解后,密切观察降解情况,每天至少定时观察6次,记录样品外观形态变化,记录样品完全溶解的时间作为改性胶原蛋白的体外降解时间。
细胞毒
按照GB/T 16886的要求,采用CCK8法对样品细胞毒进行检测。使用高密度聚乙烯塑料颗粒,用同样的浸提液,按照0.1g颗粒/ml浸提液的比例进行浸提作为细胞毒阴性对照;采用苯酚溶液作为阳性对照。每组样品取三个进行前处理,以浸提液进行细胞毒检测。以细胞相对增值率为结果判定依据。
细胞相对增殖率RGR=A/A0×100%。
式中,RGR-相对增殖率,100%;
A-样品组(阴性组、阳性组)吸光度;
A0-空白对照组吸光度。
吸光度(用以评价改性胶原蛋白的交联度)
采用2,4,6-三硝基苯磺酸(TNBS)与氨基酸残基发生反应,胶原蛋白内部交联的产生是氨基酸残基之间通过氨基和羧基发生脱水缩合,没有发生交联的氨基酸残基能够与TNBS发生反应。反应产物在367nm处产生特征吸收。通过紫外吸光光度法分析能够确定样品中自由氨基的相对数量。交联度越高,自由氨基数量越少,吸光度越低。
制备实施例
实验组A1:将10g交联促进剂PEG800添加到90g胶原蛋白原液(其中胶原蛋白的含量为0.5%(m/m))中,辐射剂量为15kGy下采用钴-60辐射处理60min;将辐照所得产物在冻干机中在0~-30℃下冷冻干燥140min,得到改性胶原蛋白A1。
实验组A2:同实验组A1,区别在于:交联促进剂为PEG2000,且使用前用水浴将PEG2000融化。
实验组A3:同实验组A1,区别在于:交联促进剂为乙二醇甲醚。
实验组A4:同实验组A1,区别在于:交联促进剂为丙三醇。
对照组C1(空白对照):同实验组A1,区别在于:未添加任何交联促进剂。
对照组C2(未交联):90g胶原蛋白原液(其中胶原蛋白的含量为0.5%(m/m))中加纯化水10g,然后于冻干机中在0~-30℃下冷冻干燥140min,得到未经交联的胶原蛋白海绵。
对照组C3(甲醛交联):超滤除菌100g胶原蛋白原液(其中胶原蛋白的含量为0.5%),接着将其无菌操作注入到冻干盘中在0~-30℃下进行冻干140min,再将冻干品在浓度为40%的甲醛蒸汽中进行交联和固定240min。
实验组A1-A4和对照组C3的具体的技术路线如图1所示。
实验组A5:将10g交联促进剂PEG800添加到90g胶原蛋白原液(其中胶原蛋白的含量为2.4wt%)中,将所得混合物在辐射剂量为25kGy下采用钴-60辐射处理60min;将辐照所得产物在冻干机中在0~-30℃下冷冻干燥140min,得到改性胶原蛋白A5。
实验组A6:同实验组A5,区别在于:交联促进剂为PEG2000,且使用前用水浴将PEG2000融化。
实验组A7:同实验组A5,区别在于:交联促进剂为乙二醇甲醚。
实验组A8:同实验组A5,区别在于:交联促进剂为丙三醇。
对照组C4(空白对照):同实验组A5,区别在于:未添加任何交联促进剂。
对照组C5(未交联):90g胶原蛋白原液(其中胶原蛋白的含量为2.4wt%)中加纯化水10g,然后于冻干机中在0~-30℃下冷冻干燥140min,得到未经交联的胶原蛋白海绵。
对照组C6(甲醛交联):超滤除菌100g胶原蛋白原液(其中胶原蛋白的含量为2.4wt%),接着将其无菌操作注入到冻干盘中在0~-30℃下进行冻干140min,再将冻干品在浓度为40%的甲醛蒸汽中进行交联和固定240min。
对照组C7:同对照组C1,区别在于:辐照剂量为3kGy。
对照组C8:同对照组C1,区别在于:辐照剂量为40kGy。
检测实施例
分别对实验组A1-A8和对照组C1-C8所得产物进行微生物含量、吸水量、拉伸强度、体外降解时间、细胞毒性和吸光度检测。
表1为实验组A1-A8和对照组C1-C8所得产物的微生物含量测试结果。
表1
从表1可以看出:样品初始污染菌的检测结果均<1CFU/件(即1CFU/0.1g)。根据ISO 11137-2中辐照灭菌剂量的要求,达到15kGy的辐射剂量即可达到灭菌目的。在无菌检验后,阴性管无菌生长,阳性管正常生长,样品管无菌生长。无菌检验也表明通过辐照交联后,样品的微生物被全部杀灭,达到10-6SAL(无菌保证水平),尽管在低于15kGy的辐射剂量依然可以满足样品交联的要求,但同时未达到灭菌,因此辐射剂量至少应不小于15kGy。C7样品接受辐照剂量为3kGy,无法达到10-6SAL(无菌保证水平)灭菌水平。
表2为实验组A1-A8和对照组C1-C8所得产物的吸水量测试结果。
表2
从表2可以看出:未交联的胶原蛋白海绵在水中一定时间后溶解,相比而言,辐照交联的样品在吸水率方面更为优异;在辐照剂量较低的情况下,胶原蛋白的交联效果不理想,辐照剂量不足以将其交联;在辐照剂量过高时,在样品进行交联的同时出现降解且降解效果大于交联效果,导致最终成品的结构中无法形成稳定的交联结构。
对于应用到对人体有体液渗出护理的生物医学材料方面而言,采用本发明所述改性方法处理可有效提高所得改性胶原蛋白的稳定性,因此更加有利于应用。
此外,发明人进一步发现:增加胶原蛋白浓度也能够显著增加样品最终的吸水量,单位重量的材料吸水效率会更强。
表3为实验组A1-A8和对照组C1-C8所得产物的拉伸强度测试结果。
表3
从表3可以看出:相比于未进行交联处理和通过现有甲醛交联进行处理,本发明所述辐照交联处理对所得样品的拉升强度的提升效果更显著,从而可更大程度地增强胶原蛋白材料的物理性能。
表4为实验组A1-A8和对照组C1-C8所得产物的体外降解测试结果。
表4
从表4可以看出:未交联样品十分容易被降解,这与其交联程度低有直接关系,甲醛交联的样品相对较好,辐照交联的样品体外降解时间较长,添加具有富羟基的交联促进剂之后,样品的体外降解时间能够进一步延长。此外,可以通过调节交联促进剂的比例来调节体外降解的时间。本试验通过采用2.4%浓度的胶原蛋白进行试验,体外降解的趋势和0.5%胶原蛋白的样品一致,通过改变胶原蛋白和乙二醇甲醚的比例能够较好地控制体外降解时间。辐照剂量过低时交联效果不足,导致样品容易降解,而辐照剂量过高又会导致降解效果大于交联效果,样品同样容易出现降解。
表5为实验组A1-A8和对照组C1-C8所得产物的细胞毒测试结果。
表5
从表5可以看出:采用传统化学交联剂甲醛交联的样品其表现出较强的细胞毒性,未交联胶原蛋白海绵以及辐照交联的样品细胞毒性均小于2级,符合人体生物材料生物学评价要求。本试验辐照交联样品采用胶原蛋白含量为2.4%的样品最终检测细胞毒<2级。添加交联促进剂以及不添加的样品其细胞毒均<2级。因乙二醇甲醚自身存在一定的毒性,通过本发明交联方式可显著性降低其细胞毒性。
因此,在添加富羟基的交联促进剂时进行辐照交联能够使得所得生物材料的细胞毒符合人体植入生物材料的要求。
表6为实验组A1-A8和对照组C1-C8所得产物的吸光度测试结果。
表6
从表6可以看出:未交联样品交联程度最低,这也导致其结构更为松散,甲醛交联的样品交联度较高,辐照交联的样品交联度也较高,辐照能够有效地将样品的交联度提升,乙二醇甲醚对于交联后样品的交联度提升程度最大。
因此,采用本发明所述方法改性所得材料有应用的潜力,在样品胶原浓度处在0.5%~2.4wt%的范围内均能得到交联度良好的材料。
综上所述,本发明提供了一种新型的可有效改善胶原蛋白稳定性的方法,所述方法可在较低的辐照剂量下高效改善胶原蛋白的交联度,从而显著提升胶原蛋白的稳定性。不仅如此,所述方法还可有效提升胶原蛋白的吸水性能、力学性能,在不引入任何有毒物质的情况下实现同步杀菌效果。
在本发明提及的所有文献都在本申请中引用作为参考,就如同每一篇文献被单独引用作为参考那样。此外应理解,在阅读了本发明的上述讲授内容之后,本领域技术人员可以对本发明作各种改动或修改,这些等价形式同样落于本申请所附权利要求书所限定的范围。
- 还没有人留言评论。精彩留言会获得点赞!