糖簇化合物及其药学上可接受的盐、它们的用途和一种药物组合物的制作方法
本申请涉及药物化学
技术领域:
,更具体地说,涉及一种透明质酸寡糖糖簇化合物及其药学上可接受的盐、透明质酸寡糖糖簇化合物或其药学上可接受的盐的用途、和包含透明质酸寡糖糖簇化合物或其药学上可接受的盐的药物组合物。
背景技术:
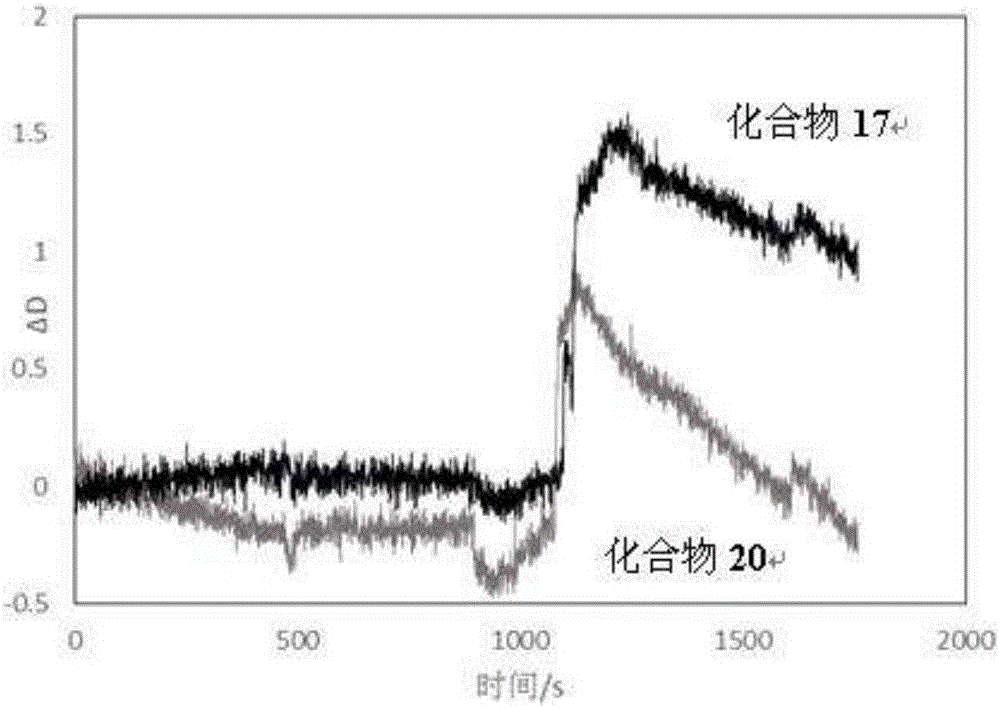
:在种类繁多的多糖中,有一类杂多糖被称为糖胺聚糖(glycosaminoglycan),也曾称粘多糖(mucopolysaccharide),其为不分支的长链聚合物,由含己糖醛酸(角质素除外)和己糖胺成分的重复复合单位构成。糖胺聚糖主要有透明质酸、硫酸软骨素、硫酸皮肤素、硫酸角质素和肝素等,它们与生物体内很多生理过程密切联系,因此对糖胺聚糖的结构、性质、以及功能化进行研究具有重大的科学和应用价值。其中,透明质酸(hyaluronicacid、hyaluronan、HA)作为非常重要的一员,具有多样的生理功能和优良的物化性质,而且作为人体中无处不在的免疫性多糖,对许多细胞和组织功能至关重要。透明质酸是一种直链聚阴离子多糖,由(1→4)-β-D-葡萄糖醛酸和(1→3)-β-D-N-乙酰氨基葡萄糖双糖重复单元组成,其结构如下所示:透明质酸广泛存在于动物的各种组织中,具有特殊的生理功能。透明质酸除保水作用外还具有调节渗透压、维持组织形态、屏障扩散、润滑关节和缓冲应力等生理功能。更为重要的是,透明质酸能够被特定的细胞受体,如CD44,识别从而调控细胞的粘附、生长、分化和激活特异性细胞类型,调控免疫反应、血管化和愈合过程。近年来,以合成或天然聚合物为载体,通过共价结合治疗药物制备共价轭合物来延长药物半衰期或降低其毒副作用的研究已引起国内外的广泛关注。其中透明质酸以其优良特性成为了制备轭合物的首选载体:①透明质酸具有良好的生物降解性、生物相容性、无毒、无抗原性,在体内可酶解消除;②透明质酸分子中含有大量羧基、羟基和N-乙酰氨基,这些基团均可被化学修饰,尤其是羧基易与小分子药物结合,并保持较高的载药率;③透明质酸与药物共价结合能够延长药物的半衰期、提高稳定性、降低免疫原性和提高药物溶解性;④透明质酸与其特异性受体CD44的专一性结合使HA药物轭合物具备药物增稠、靶向治疗作用,同时也能达到缓释、控释的目的。目前,透明质酸及其衍生物已被广泛用作多种抗癌药物、多肽和蛋白类药物的载体,实验证实制备成HA药物轭合物之后能够延长药物在用药部位的存留时间,提高生物利用度,减少给药次数,降低不良反应。然而透明质酸作为药物载体也存在一些问题:①透明质酸为高聚物,其分子量存在不均一性,因此其与药物的缀合物的连接位点、载药量并不是精确确定的;②不同分子量的透明质酸对CD44的作用并不相同,且机理尚未确定。技术实现要素:本申请的发明人采用结构确定的透明质酸寡糖组装成糖簇,利用其糖簇效应,既保证了其活性,又克服了其诸多不足。本申请的第一个目的是提供一种透明质酸寡糖糖簇化合物或其药学上可接受的盐。本申请的第二个目的是提供上述化合物或其药学上可接受的盐的用途。本申请的第三个目的是提供包含上述化合物或其药学上可接受的盐的药物组合物。具体地说,本申请以透明质酸寡糖(四糖、六糖等)为起始原料,采用多功能基的分子(如多元醇、多元胺、树枝状大分子)作为分子骨架,合成了一系列具有式I的基于透明质酸寡糖的糖簇。本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,或其药学上可接受的盐这里,式I中,m为2-4中的整数,优选地,m=2或3;n为2-15中的整数,优选地,n为2、3、4、5、6、8、或9;L1为与糖直接相连的连接臂-1,选自直链或支链C1-C8烷烃亚基、氧代的直链或支链C1-C8烷烃亚基、C3-C8环烷烃亚基、或聚乙二醇亚基,优选地,为聚乙二醇亚基,更优选地,为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基、硫桥键、-HN-C(O)-或-(O)C-NH-的酰胺键结构,更优选地,为三氮唑基;L2为连接L1和T的连接臂-2,选自直链或支链C1-C8烷烃亚基、氧代的直链或支链C1-C8烷烃亚基、C3-C8环烷烃、或聚乙二醇亚基;优选地,亚基为-CH2-、-CH2CH2-O-CH2CH2-O-CH2CH2-或-CH2CH2CH2C(O)-;Y为糖簇的多官能团支架,即带有多羟基或多氨基的树枝状大分子化合物基团;优选地,选自2-叔丁氧羰酰氨基-1,3-丙二醇、甘油、季戊四醇、没食子酰丙胺或聚酰胺PAMAM。在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=2;n=2;L1为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基;L2为-CH2-;Y为2-叔丁氧羰酰氨基-1,3-丙二醇,即:在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=2;n=3;L1为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基;L2为-CH2-;Y为甘油,即:在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=2;n=4;L1为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基;L2为-CH2-;Y为季戊四醇,即:在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=2;n=8;L1为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基;L2为-CH2CH2CH2C(O)-;Y为G1-PAMAM,即:在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=2;n=9;L1为-CH2-;T为三氮唑基;L2为-CH2CH2-O-CH2CH2-O-CH2CH2-;Y为没食子酸衍生物,即:在本申请的优选实施方案中,本申请提供了一种如式I所示的透明质酸寡糖糖簇化合物,其中,m=3;n=3;L1为-CH2CH2-O-CH2CH2-O-CH2CH2-;T为三氮唑基;L2为-CH2-;Y为甘油。本申请还提供了一种透明质酸寡糖糖簇化合物或其药学上可接受的盐,其中,所述化合物选自化合物10、化合物13、化合物17、化合物20、化合物23和化合物32。本申请所提供的透明质酸寡糖糖簇化合物的药学上可接受的盐,选自酸加成盐或碱金属盐;所述酸加成盐选自无机酸盐或有机酸盐;所述无机酸盐选自氢卤酸盐(如盐酸盐、氢溴酸盐、或氢碘酸盐等)、硫酸盐、硫酸氢盐、或磷酸盐等,优选地为盐酸盐;所述有机酸盐选自甲磺酸盐、苯磺酸盐、对甲苯磺酸盐、马来酸盐、富马酸盐、琥珀酸盐、枸橼酸盐、或苹果酸盐等;所述碱金属盐选自钾盐、钠盐或锂盐。在本申请的实施方案中,本申请提供的基于透明质酸寡糖的糖簇,其中,原料透明质酸寡糖既可以是商品化的试剂,也可以是通过寡糖合成方法合成的产物,更可以是通过酶降解高分子量透明质酸后分离得到的产物。本申请也提供了上述如式I所示的透明质酸寡糖糖簇化合物或其药学上可接受的盐作为抗肿瘤药物或者作为药用靶向辅料(例如基因药物载体)的用途。透明质酸为高分子量聚糖,其粘性较高,成药性较差,而本申请的透明质酸寡糖糖簇在分子量相差数十倍的情况可以有效地克服这一缺点;不仅与CD44蛋白具有较好的结合力,而且对CD44高表达肿瘤细胞具有一定的结合能力,且具有抑制肿瘤细胞迁移的作用。本申请又提供了包含上述如式I所示的透明质酸寡糖糖簇化合物或其药学上可接受的盐的药物组合物,其中,所述透明质酸寡糖糖簇化合物作为药物组合物的活性成分、或者药用靶向辅料(例如基因药物载体)。附图说明图1表示的是本申请透明质酸寡糖糖簇化合物23和化合物13的石英晶体微天平结果。图2表示的是本申请透明质酸寡糖糖簇化合物20和化合物17的石英晶体微天平结果。图3表示的是对照组划痕实验结果。图4表示的是本申请透明质酸寡糖糖簇化合物17划痕实验结果。图5表示的是高分子量透明质酸划痕实验结果。具体实施方式下面通过实施例来进一步地说明本申请的可实施性,并非对本申请保护范围的限制。检测仪器:核磁:BrukerAV-400型核磁共振仪,溶剂为CDCl3,TMS为内标质谱:BrukerAPEXIV型质谱仪实施例1步骤(1):(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-1,4,6-三-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺2的合成将10.0g(β-D-吡喃葡萄糖醛酸)-(1→3)-(2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(β-D-吡喃葡萄糖醛酸)-(1→3)-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺1溶于1.2L浓度为0.08M的冷氯化氢甲醇溶液中。而后将反应液于4℃搅拌96h。反应结束后将反应液用三乙胺中和至中性,而后减压蒸馏,并浓缩至干。加入吡啶50mL,冷却至0℃,缓慢滴加乙酸酐25mL后将反应液缓慢升至室温,并于室温下搅拌24h。待反应结束后,将反应液在冰水浴中冷却,缓慢滴加甲醇30mL,并于该温度下搅拌1h,将反应液减压蒸馏后溶于300mL乙酸乙酯中,有机相依次用50mL浓度为1M的盐酸洗涤3次,水相用100mL乙酸乙酯萃取三次。合并有机相,并依次用60mL饱和碳酸氢钠水溶液洗涤2次,60mL饱和食盐水洗涤1次,将有机相用无水硫酸钠干燥,过滤,减压蒸馏,所得产品用二氯甲烷:乙酸乙酯:甲醇=1:1:0.14(洗脱剂含0.1%三乙胺)快速硅胶柱层析纯化,得14.8g白色固体。总收率93.8%。HRMS(ESI):[M+H]+计算值1225.3777,实测值[M+H]+:1225.3750。步骤(2):(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺3的合成取10.0g(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-1,4,6-三-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺溶于100mL分析纯四氢呋喃中,在搅拌下缓慢滴加6mL3-N,N-二甲基-氨基丙胺,并于室温下搅拌3h。将反应液用200mL氯仿稀释,有机相用60mL浓度为1M的盐酸溶液洗涤3次,水相用100mL氯仿萃取3次,合并有机相,用100mL饱和碳酸氢钠水溶液洗涤1次,60mL饱和食盐水洗涤1次,有机相用无水硫酸钠干燥,过滤,减压浓缩得8.5g淡黄色粉末,收率88%。HRMS(ESI):[M+H]+计算值1182.3599,实测值[M+H]+:1182.3658。步骤(3):O-(((2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-)-O-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺基)三氯乙酰亚胺酯4的合成取干燥的(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1-3)-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺粗品7.0g(6.0mmol)溶于60mL干燥二氯甲烷中,加入三氯乙腈6.7mL(60mmol),将此混合物在氩气气氛下于冰水浴中冷却。之后缓慢滴加1,8-二氮杂二环十一碳-7-烯149μL(0.9mmol)。反应液于0℃下搅拌2.5h。待反应完成后将反应液减压浓缩,并用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=3:1:0.10,含0.1%三乙胺)得5.5g白色无定形固体,收率79%。1HNMR(400MHz,CDCl3)δ8.83(s,1H,C=NH),6.23(d,J=3.7Hz,1H),5.84(d,J=7.5Hz,1H),5.77(d,J=9.6Hz,1H),5.21(dd,J=J=9.3Hz,1H),5.15-5.07(m,3H),4.90(dd,J=J=9.4Hz,1H),4.83-4.73(m,4H),4.63(d,J=7.8Hz,1H),4.59(ddd,J=10.1Hz,3.8Hz,3.8Hz,1H),4.43(dd,J=J=9.7Hz,1H),4.29(dd,J=4.2Hz,12.4Hz,1H),4.18(dd,J=4.2Hz,12.5Hz,1H),4.12-3.85(m,11H),3.72(s,3H),3.66-3.64(m,1H),3.14(ddd,J=17.4Hz,8.0Hz,8.0Hz,1H),2.08-2.01(m,33H,COCH3)。13C(100MHz,CDCl3),171.0,170.8,170.2,170.1,169.8,169.8,169.7,169.4,169.2,169.1,167.9,167.0,160.2,100.9,100.0,98.7,95.3,90.8,77.3,76.8,75.8,74.9,72.3,72.1,71.8,71.8,71.7,71.6,70.2,69.4,68.0,67.7,61.8,61.7,57.7,53.0,52.8,51.8,23.6,23.2,20.8,20.7,20.6,20.6,20.5,20.5。HRMS(ESI):[M+H]+计算值1326.2768,实测值[M+H]+:1326.2738。步骤(4):O-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-2-甲基-(4,6-二-O-乙酰基-1,2-二脱氧-α-D-吡喃葡萄糖)[2,1-d]2-噁唑啉5的合成。取干燥的(((2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-)-O-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺基)三氯乙酰亚胺酯9.0g(6.8mmol)溶于80mL干燥二氯甲烷中,并于氩气气氛下在冰水浴中冷却。缓慢滴加三氟甲磺酸三甲基硅酯182μL(1.02mmol)。反应液于0℃下搅拌0.5h。待反应完成后滴加150μL三乙胺淬灭反应。将反应液减压浓缩至干,并用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=3:1:0.15,含0.1%三乙胺)得7.1g白色无定形固体,收率90%。1HNMR(400MHz,CDCl3)δ5.92(d,J=6.6Hz,1H),5.82(d,J=7.3Hz,1H),5.20(dd,J=J=9.3Hz,1H),5.12(dd,J=J=9.0Hz,1H),5.06(dd,J=J=9.2Hz,1H),4.92-4.78(m,6H),4.65(d,J=7.8Hz,1H),4.60(d,J=7.8Hz,1H),4.52(m,1H),4.41-4.30(m,2H),4.20(dd,J=4.2Hz,12.5Hz,1H),4.12-3.92(m,6H),3.82(s,3H),3.71-3.63(m,11H),3.41(m,2H),3.30(m,1H),3.01(m,1H),2.06-1.96(m,33H).13C(100MHz,CDCl3),171.2(2C),170.8,170.2,170.1,169.8,169.8,169.7,169.4,169.2,169.1,167.9,167.0,100.7,100.0,99.4,98.3,77.9,76.7,75.6,74.5,72.3,72.2,72.2,71.9,71.8,71.7,71.6,70.4,69.9,69.4,68.9,68.5,68.1,62.4,62.0,58.1,57.5,52.9,52.7,50.7,23.6,23.4,20.8,20.7,20.6,20.6,20.5,20.5;HRMS:计算值[M+H]+:1165.3566,实测值[M+H]+:1165.3561。步骤(5):1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷7的合成。取干燥的O-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-2-甲基-(4,6-二-O-乙酰基-1,2-二脱氧-α-D-吡喃葡萄糖)[2,1-d]2-噁唑啉5.0g溶于35mL干燥的三氯甲烷中,加入7.5g1-叠氮-2,4-二氧-6己醇6和无水氯化铜631mg。反应液于下回流10h后加入10mL蒸馏水并继续回流1h。将反应液于室温下冷却,加入饱和碳酸氢钠水溶液并剧烈搅拌。水相用10mL二氯甲烷萃取3次,合并有机相,用饱和NaCl水溶液洗涤,无水硫酸钠干燥,过滤,浓缩得无色糖浆。将该糖浆分散于30mL无水乙醚中剧烈搅拌12h,可见大量白色固体析出,过滤收集固体,将固体用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=1:1:0.10,含0.1%三乙胺)得4.84g白色无定形固体,收率84%。1HNMR(400MHz,CDCl3)δ6.08(m,2H,NHAc),5.20(dd,J=J=9.3Hz,1H),5.12(dd,J=J=9.0Hz,1H),5.06(dd,J=J=9.2Hz,1H),4.92-4.78(m,6H),4.65(d,J=7.8Hz,1H),4.60(d,J=7.8Hz,1H),4.52(m,1H),4.41-4.30(m,2H),4.20(dd,J=4.2Hz,12.5Hz,1H),4.12-3.92(m,6H),3.82(s,3H),3.71-3.63(m,11H),3.41(m,2H),3.30(m,1H),3.01(m,1H),2.06-1.96(m,33H)。13C(100MHz,CDCl3),171.2(2C),170.8,170.2,170.1,169.8,169.8,169.7,169.4,169.2,169.1,167.9,167.0,100.7,100.0,99.4,98.3,77.9,76.7,75.6,74.5,72.3,72.2,72.2,71.9,71.8,71.7,71.6,70.4,69.9,69.4,68.9,68.5,68.1,62.4,62.0,58.1,57.5,52.9,52.7,50.7,23.6,23.4,20.8,20.7,20.6,20.6,20.5,20.5。HRMS:计算值[M+H]+:1339.4450,实测值[M+H]+:1339.4482。步骤(6):全保护糖簇9的合成取10.3mg1,2,3-三(炔丙基氧)丙烷8,加入1.6mL甲醇,0.2mL去离子水,加入221.1mg1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷7,2.7mg无水硫酸铜,32.7mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入10mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得145.3mg白色固体,收率91.0%。HRMS:计算值[M+H]+:4225.4326,实测值[M+H]+:4225.5108步骤(7):糖簇化合物10的合成取100.0mg全保护糖簇9溶于5mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入2.0mL1MLiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得70.1mg白色固体,收率100%。HRMS:计算值[M+H]+:3007.0535,实测值[M+H]+:3007.0312实施例2步骤(1)至步骤(5)同实例1。步骤(6):全保护糖簇12的合成取109.1mg八己炔酰G-2PAMAM即化合物11加入4mL甲醇,0.5mL去离子水,加入643.3mg1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷7,7.7mg无水硫酸铜,95.1mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入20mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得563.3mg白色固体,收率89.3%。HRMS:计算值[M+H]+:12901.9322,实测值[M+H]+:12902.1628步骤(7):糖簇化合物13的合成取100.0mg全保护糖簇12溶于5mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入2.0mL1MLiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得76.4mg白色固体,收率100%。HRMS:计算值[M+H]+:9651.9177,实测值[M+H]+:9651.8161。这里,八己炔酰G-1PAMAM即化合物11的合成取143.2mgG1-PAMAM溶于2mL二氯甲烷中,滴入一滴N,N-二甲基甲酰胺,加入177.8mg5-己炔酸,184.0mg1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐,130.0mg1-羟基苯并三唑,室温搅拌24h。将溶剂蒸干,加入3mL甲醇溶解,以甲醇为洗脱剂过LH-20凝胶柱,旋干得205.7mg浅绿色油状液体,收率94.2%。HRMS:计算值[M+H]+:2184.3744,实测值[M+H]+:2184.3046实施例3步骤(1)至步骤(4)同实例1。步骤(5):3-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]-1-丙炔14的合成取干燥的O-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-2-甲基-(4,6-二-O-乙酰基-1,2-二脱氧-α-D-吡喃葡萄糖)[2,1-d]2-噁唑啉5.0g溶于35mL干燥的三氯甲烷中,加入2.5g炔丙醇和无水氯化铜631mg。反应液于下回流10h后加入10mL蒸馏水并继续回流1h。将反应液于室温下冷却,加入饱和碳酸氢钠水溶液并剧烈搅拌。水相用10mL二氯甲烷萃取3次,合并有机相,用饱和NaCl水溶液洗涤,无水硫酸钠干燥,过滤,浓缩得无色糖浆。将该糖浆分散于30mL无水乙醚中剧烈搅拌12h,可见大量白色固体析出,过滤收集固体,将固体用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=1:1:0.10,含0.1%三乙胺)得4.07g白色固体,收率78.2%。HRMS:计算值[M+H]+:1220.3755,实测值1220.3582步骤(6):全保护糖簇16的合成取126.9mg化合物15,加入4mL甲醇,0.5mL去离子水,加入604.4mg3-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]-1-丙炔即化合物14,7.2mg无水硫酸铜,98.1mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入20mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得598.3mg白色固体,收率96.9%。HRMS:计算值[M+H]+:13522.7516,实测值13523.0645步骤(7):糖簇化合物17的合成取100.0mg全保护糖簇16溶于5mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入2.0mL1MLiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得72.8mg白色固体,收率100%。HRMS:计算值[M+H]+:9866.6108,实测值9866.8904实施例4步骤(1)至步骤(5)同实例1。步骤(6):全保护糖簇19的合成取26.7mg2-叔丁氧羰基氨基-1,3-二(炔丙基氧)丙烷即化合物18,加入1.6mL甲醇,0.2mL去离子水,加入321.6mg1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷即化合物7,3.8mg无水硫酸铜,47.5mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入10mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得280.1mg白色固体,收率95.0%。HRMS:计算值[M+H]+:2947.0404,实测值[M+H]+:2947.0564步骤(7):糖簇化合物20的合成取100.0mg全保护糖簇19溶于5mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入2.0mL浓度为1M的LiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得72.4mg白色固体,收率100%。HRMS:计算值[M+H]+:2133.7843,实测值2133.7658实施例5步骤(1)至步骤(5)同实例1。步骤(6):全保护糖簇22的合成取28.8mg1,3-二(炔丙基氧)-2,2-二(炔丙基氧基甲基)丙烷即化合物21,加入1.6mL甲醇,0.2mL去离子水,加入643.3mg1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷即化合物7,7.6mg无水硫酸铜,95.0mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入10mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得553.1mg白色固体,收率97.9%。HRMS:计算值[M+H]+:5647.9288,实测值[M+H]+:5647.9256步骤(7):糖簇化合物23的合成取100.0mg全保护糖簇22溶于10mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入4.0mL1MLiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得71.2mg白色固体,收率100%。HRMS:计算值[M+H]+:4022.4140,实测值4022.4195实施例6步骤(1):(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-1,4,6-三-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺26的合成将10.0g(β-D-吡喃葡萄糖醛酸)-(1→3)-(2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(β-D-吡喃葡萄糖醛酸)-(1→3)-(2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(β-D-吡喃葡萄糖醛酸)-(1→3)-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺25溶于1.5L,0.08M的冷氯化氢甲醇溶液中。而后将反应液于4℃搅拌96h。反应结束后将反应液用三乙胺中和至中性,而后减压蒸馏,并浓缩至干。加入吡啶75mL,冷却至0℃,缓慢滴加乙酸酐35mL后将反应液缓慢升至室温,并于室温下搅拌24h。待反应结束后,将反应液在冰水浴中冷却,缓慢滴加甲醇45mL,并于该温度下搅拌1h,将反应液减压蒸馏后溶于450mL乙酸乙酯中,有机相依次用75mL的1M盐酸洗涤3次,水相用150mL乙酸乙酯萃取三次。合并有机相,并依次用90mL饱和碳酸氢钠水溶液洗涤2次,90mL饱和食盐水洗涤1次,将有机相用无水硫酸钠干燥,过滤,减压蒸馏,所得产品用二氯甲烷:乙酸乙酯:甲醇=1:1:0.14(洗脱剂含0.1%三乙胺)快速硅胶柱层析纯化,得15.1g白色固体。总收率98.0%。HRMS(ESI):[M+H]+计算值1785.5398,实测值[M+H]+:1785.5406。步骤(2):(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺27的合成取10.0g(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1-4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1-3)-1,4,6-三-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺26溶于100mL分析纯四氢呋喃中,在搅拌下缓慢滴加6mL3-N,N-二甲基-氨基丙胺,并于室温下搅拌3h。将反应液用200mL氯仿稀释,有机相用60mL的1M盐酸溶液洗涤3次,水相用100mL氯仿萃取3次,合并有机相,用100mL饱和碳酸氢钠水溶液洗涤1次,60mL饱和食盐水洗涤1次,有机相用无水硫酸钠干燥,过滤,减压浓缩得8.8g淡黄色粉末,收率90.2%。HRMS(ESI):[M+H]+计算值1743.5292,实测值[M+H]+:1743.5246。步骤(3):O-(((2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-)-O-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺基)三氯乙酰亚胺酯28的合成取干燥的(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-吡喃葡萄糖醛酸甲酯)-(1→3)-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-吡喃葡萄糖胺277.0g(6.0mmol)溶于60mL干燥二氯甲烷中,加入三氯乙腈6.7mL(60mmol),将此混合物在氩气气氛下于冰水浴中冷却。之后缓慢滴加1,8-二氮杂二环十一碳-7-烯149μL(0.9mmol)。反应液于0℃下搅拌2.5h。待反应完成后将反应液减压浓缩,并用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=3:1:0.10,含0.1%三乙胺)得5.8g白色无定形固体,收率76.5%。HRMS(ESI):[M+H]+计算值1886.4389,实测值[M+H]+:1886.4405。步骤(4):O-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-2-甲基-(4,6-二-O-乙酰基-1,2-二脱氧-α-D-吡喃葡萄糖)[2,1-d]2-噁唑啉29的合成。取9.0g(6.8mmol)干燥的O-(((2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-葡萄糖胺)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-)-O-4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-D-葡萄糖胺基)三氯乙酰亚胺酯28溶于80mL干燥二氯甲烷中,并于氩气气氛下在冰水浴中冷却。缓慢滴加三氟甲磺酸三甲基硅酯182μL(1.02mmol)。反应液于0℃下搅拌0.5h。待反应完成后滴加150μL三乙胺淬灭反应。将反应液减压浓缩至干,并用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=3:1:0.15,含0.1%三乙胺)得7.2g白色无定形固体,收率87.8%。HRMS:计算值[M+H]+:1725.5187,实测值[M+H]+:1725.5165。步骤(5):1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷30的合成。取5.0g干燥的O-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-2-甲基-(4,6-二-O-乙酰基-1,2-二脱氧-α-D-吡喃葡萄糖)[2,1-d]2-噁唑啉29溶于35mL干燥的三氯甲烷中,加入7.5g1-叠氮-2,4-二氧-6己醇6和无水氯化铜631mg。反应液于下回流10h后加入10mL蒸馏水并继续回流1h。将反应液于室温下冷却,加入饱和碳酸氢钠水溶液并剧烈搅拌。水相用10mL二氯甲烷萃取3次,合并有机相,用饱和NaCl水溶液洗涤,无水硫酸钠干燥,过滤,浓缩得无色糖浆。将该糖浆分散于30mL无水乙醚中剧烈搅拌12h,可见大量白色固体析出,过滤收集固体,将固体用硅胶快速柱色谱纯化(二氯甲烷:乙酸乙酯:甲醇=1:1:0.10,含0.1%三乙胺)得4.7g白色无定形固体,收率85.5%。HRMS:计算值[M+H]+:1725.5187,实测值[M+H]+:1725.5165。步骤(6):全保护糖簇31的合成取10.3mg1,2,3-三(炔丙基氧)丙烷8,加入1.6mL甲醇,0.2mL去离子水,加入342.3mg1-叠氮-2,4-二氧-6-O-[(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-β-D-吡喃葡萄糖基)-(1→4)-(2,3,4-三-O-乙酰基-β-D-葡萄糖醛酸甲酯基)-(1→3)-(4,6-二-O-乙酰基-2-脱氧-2-N-乙酰氨基-α-D-吡喃葡萄糖基)]己烷30,2.7mg无水硫酸铜,32.7mg抗坏血酸钠,室温搅拌反应8h。将溶剂蒸干,加入10mL甲醇,滤去不溶物,以甲醇为洗脱剂过LH-20凝胶柱,旋干得285.0mg白色固体,收率96.3%。HRMS:计算值[M+H]+:5909.9441,实测值[M+H]+:5909.9356步骤(7):糖簇化合物32的合成取100.0mg全保护糖簇31溶于5mL四氢呋喃、于冰盐浴中冷却,然后缓慢加入5.0mL1MLiOH水溶液、30%H2O2水溶液,滴加完毕后于室温下反应12h,待反应完成后用“Amberlite”IR-120氧离子交换树脂中和反应液至中性。过滤去除树脂,将滤液冷冻干燥,并在盛有无水氯化钙的干燥器中干燥,得70.1mg白色固体,收率100%。HRMS:计算值[M+H]+:4144.3879,实测值[M+H]+:4144.5616效果例1表面等离子共振(SPR)测定本申请透明质酸寡糖糖簇化合物与透明质酸受体CD44蛋白的结合能力将CD44(R&D,RecombinanthumanCD44-Fc)固定于BiacoreT200的CM7芯片上。芯片先由EDC:NHS=4:1活化液活化7min,CD44溶于10mMpH=4醋酸盐缓冲液配置成17μg/mL并流经芯片5min。后由pH=8.51M乙醇胺溶液封闭多余活性基团。将本申请的糖簇化合物13、17、20、23配制成一定浓度的工作溶液,在BiacoreT200仪器上测定其与CD44结合能力。化合物编号KD(μM)133.072172.493205.195239.431SPR数据表明本申请的透明质酸寡糖糖簇化合物与CD44蛋白均具有很好的结合能力。效果例2石英晶体微天平(QCM)测定透明质酸寡糖糖簇对CD44高表达肿瘤细胞的靶向性石英晶体微天平(QCM)是一种以质量变化为依据的传感器,具有高度的灵敏性。芯片(标准芯片,镀金)预处理:紫外/臭氧处理10min,浸入10mL蒸馏水,2mL双氧水,2mL氨水组成的混合溶液,75℃加热5min,后用蒸馏水冲洗,超声。用Ar气流吹干。然后浸入5mM3-巯基丙酸中反应过夜。反应完成后用乙醇洗,再用水洗,放入多聚赖氨酸中反应1h。用水洗干净晶片,用Ar气吹干。本实验采用CD44高表达的人乳腺癌细胞株MDA-MB-231(北京大学药学院)进行测定。培养基:DMEM高糖培养基,含10%血清,1%100U/ml青霉素和链霉素(双抗)。细胞悬浮于无血清培养基中。糖簇样品用无血清培养基配制,浓度为1mg/mL。所用石英晶体微天平型号为Q-SenseE4。将处理好的芯片装于流动池中,先将细胞悬液固定于芯片表明,待基线稳定后进入待测样品,流速为70μL/min。石英晶体微天平的结果(参见图1和图2)可以分析出:所合成的透明质酸寡糖糖簇,即使是最小的二价糖簇,与CD44高表达的细胞也有一定的结合能力。效果例3划痕实验测定透明质酸寡糖糖簇对迁移性肿瘤细胞迁移活动的影响透明质酸与其受体结合会影响细胞的一系列活动,其中非常重要的一项是影响肿瘤细胞的迁移。现有技术表明:较小分子量的透明质酸会促进肿瘤细胞的迁移,而高分子量的透明质酸会抑制透明质酸的迁移。将处于对数生长期的MDA-MB-231细胞以2×105个/孔接种于12孔板,每孔1ml,置于孵箱(5%CO2,37℃)中培养24h。用含1%血清的培养基配制本申请化合物17和高分子量透明质酸(MW>500kDa)浓度为100μg/ml的溶液。取出培养24h的细胞,用直尺和10μl枪头枪头在孔内划痕,弃去培养基,用无血清培养基洗两遍,加入配制好的药液,每个浓度两个孔,设置空白对照组。在孵箱中培养24h。赋育结束后,在倒置显微镜下,选取随机视野,观察细胞伤口愈合情况(参见图3-5)。由图可以看出,未经处理的对照组细胞划痕接近愈合。以本申请化合物17(分子量为10kDa左右)处理过的细胞其划痕依旧较宽,显示了化合物17具有一定的抑制肿瘤细胞迁移的作用;与分子量为500kDa以上的阳性对照高分子量透明质酸相同,也抑制该肿瘤细胞迁移的作用。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1