一种InDel位点基因型分型的方法与流程
本发明涉及分子生物学技术领域,尤其涉及一种InDel位点基因型分型的方法。
背景技术:
插入缺失(Insertion and Deletion,简称InDel)是指同源序列的比对中出现的至少1bp核苷酸的差异,这种差异称为InDels;InDel在基因组中的含量仅次于单核苷酸多态性(SNP)标记,具有数量多、分布广泛、变异丰富等特点。根据基因组中插入缺失位点,设计扩增这些插入缺失位点的PCR引物,产生的多态性位点即为插入缺失(InDel)标记。插入缺失(InDel)标记在系统发育推断、遗传诊断、药物设计等方面具有重要的应用潜力。
目前,常用的InDel分型方法主要是测序法和芯片法,测序法和芯片法实验操作繁琐、效率较低并且依赖相关大型仪器平台作为支撑,费用昂贵。
技术实现要素:
有鉴于此,本发明的目的在于提供一种成本低、操作简单灵活、准确的基因型分型的方法。
为了实现上述发明目的,本发明提供以下技术方案:
本发明提供了一种InDel位点基因型分型的方法,包括以下步骤:
1)将目的基因DNA序列进行T载体克隆测序,得到目的基因克隆产物序列,将所述目的基因克隆产物序列进行多重比对,得到InDel位点;
2)根据所述InDel位点设计PCR扩增引物,所述PCR扩增引物包括正向引物和反向引物,所述正向引物的5'末端采用荧光基团修饰;
3)用所述PCR扩增引物对待测样本全基因组DNA进行PCR扩增获得扩增产物,用毛细管荧光凝胶电泳检测扩增产物,若扩增产物中有两条条带则该InDel位点的基因型为杂合型ID;若有一条条带且片段大小与杂合型ID中大的片段相同,则该InDel位点基因型为插入纯合型II;若有一条条带且片段大小与杂合型ID中小的片段相同,则表示该InDel位点为缺失纯合型DD。
优选的,所述荧光基团为FAM或HEX。
优选的,所述多重比对采用MEGA软件中的ClustalW模块。
优选的,所述待测样本为杨木树种。
优选的,所述目的基因为尿苷二磷酸葡糖醛酸脱羧酶基因1。
优选的,所述尿苷二磷酸葡糖醛酸脱羧酶基因1上的InDel位点分别为InDel1、InDel2、InDel3、InDel4和InDel5;所述InDel1位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第401bp到409bp处,所述InDel2位于第1399bp到1401bp处,InDel3位于第1427bp到1438bp,InDel4位于第2094bp到2103bp处,InDel5位于第4143bp到4152bp。
优选的,根据InDel1设计的引物为正向引物SEQ ID NO.1和反向引物SEQ ID NO.2;根据InDel2和InDel3设计的引物为正向引物SEQ ID NO.3和反向引物SEQ ID NO.4;根据InDel4设计的引物为正向引物SEQ ID NO.5和反向引物SEQ ID NO.6;根据InDel5设计的引物为正向引物SEQ ID NO.7和反向引物SEQ ID NO.8。
优选的,步骤3)中所述PCR扩增的条件为:95℃预变性5min;95℃变性30s,53~57℃退火30s,72℃延伸30s,35个循环;最后72℃延伸5min。
优选的,所述PCR扩增用扩增体系包括以下组分:0.7μl的待测样品全基因组DNA,0.15μl的0.8UTaq酶,0.3μl 0.2mM的dNTPs,0.9μl 25mM的MgCl2,1.25μl的10×PCR buffer,0.4μl 100nmolL-1的正向引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。
优选的,所述基因组DNA的浓度为5~20ng/μl。
本发明的有益效果:本发明提供的InDel位点基因型分型的方法通过多重比对获得目的基因InDel位点,根据InDel位点设计PCR扩增引物,包括正向引物和反向引物,采用荧光基团修饰正向引物的5’端,然后对待测样本DNA进行PCR扩增,用毛细管荧光凝胶电泳检测扩增产物确定所述待测样本中InDel位点的基因型。本发明对引物组中的正向引物5'末端采用荧光基团进行修饰,可显著地保留荧光信号强度、减少InDel位点的非特异性扩增,提高了基因型检测的准确度;同时极大地简化了实验步骤和配套条件,并显著地降低了实验成本。
进一步的,在适宜退火温度和PCR反应条件下,进一步提高荧光信号强度,提高基因型检测的准确度。
附图说明
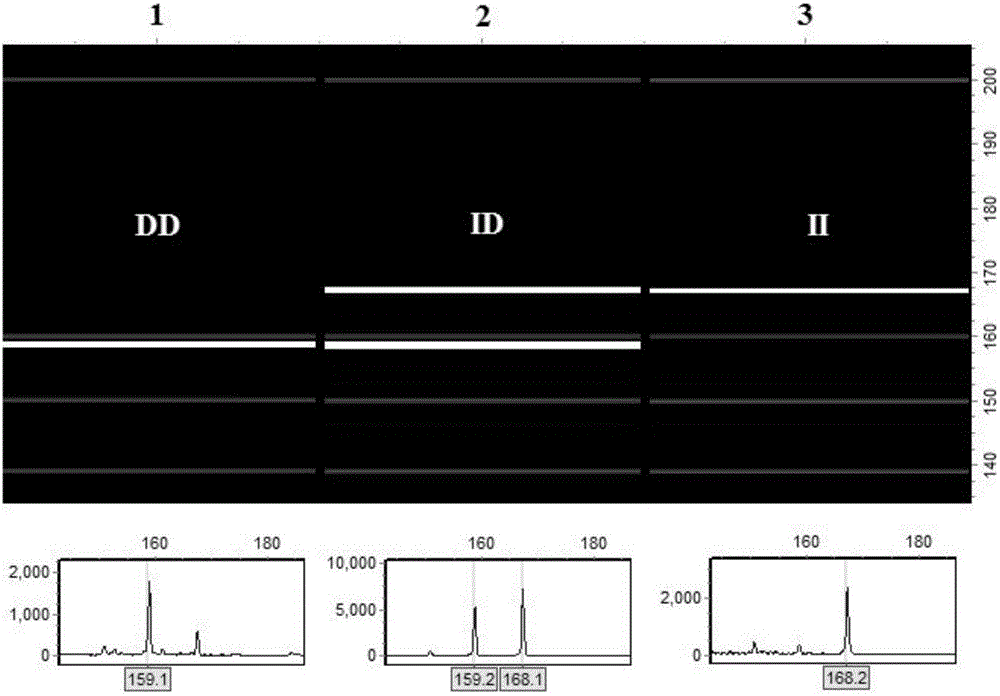
图1为实施例1中毛细管荧光凝胶电泳结果图。
具体实施方式
本发明提供了一种InDel位点基因型分型的方法,包括以下步骤:1)将目的基因DNA序列进行T载体克隆测序,得到目的基因克隆产物序列,将所述目的基因克隆产物序列进行多重比对,得到InDel位点;2)根据所述InDel位点设计PCR扩增引物,所述PCR扩增引物包括正向引物和反向引物,所述正向引物的5'末端采用荧光基团修饰;3)用所述PCR扩增引物对待测样本DNA进行PCR扩增获得扩增产物,毛细管荧光凝胶电泳检测扩增产物,若扩增产物中有两条目的条带则该InDel位点的基因型为杂合型ID;若有一条目的条带且片段大小与杂合型ID中大的片段相同,则该InDel位点基因型为插入纯合型II;若有一条目的条带且片段大小与杂合型ID中小的片段相同,则表示该InDel位点为缺失纯合型DD。
本发明提供的InDel位点基因型分型的方法适用于林木树种,优选的为杨木树种,如本发明的实施例中所用毛白杨。
本发明将目的基因DNA序列进行T载体克隆测序,得到目的基因克隆产物序列。本发明中所述的目的基因为待测样品的任一基因,优选的为尿苷二磷酸葡糖醛酸脱羧酶基因1,优选的所述尿苷二磷酸葡糖醛酸脱羧酶基因1的序列如SEQ ID NO.9所示。
在本发明选定目的基因后,将所述目的基因DNA序列进行T载体克隆测序,得到目的基因克隆产物序列。本发明对所述的T载体克隆测序的具体步骤没有特殊限定,采用本领域常规的T载体转化大肠杆菌DH52感受态细胞克隆测序方法步骤即可。
得到目的基因克隆产物序列后,本发明将所述目的基因克隆产物序列进行多重比对,得到InDel位点。在本发明中,所述的多重比对优选的采用MEGA软件中的ClustalW模块,具体的多重比对的参数优选的为软件模块中的默认参数。
具体的在本发明实施例中以尿苷二磷酸葡糖醛酸脱羧酶基因1为目的基因,对目的基因进行T载体克隆测序,后采用MEGA软件中的ClustalW模块进行多重比对,确定的InDel位点有5个,分别为InDel1、InDel2、InDel3、InDel4和InDel5;所述InDel1位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第401bp到409bp处,所述InDel2位于第1399bp到1401bp处,InDel3位于第1427bp到1438bp,InDel4位于第2094bp到2103bp处,InDel5位于第4143bp到4152bp。
本发明在获得上述InDel位点后,分别根据上述InDel位点设计引物,包括正向引物和反向引物,所述正向引物的5'末端采用荧光基团修饰。优选的根据InDel1设计的引物为正向引物SEQ ID NO.1和反向引物SEQ ID NO.2;正向引物的序列为UXS1ID1F:5'-CTCCCGCCTTAACCCATCT-3';反向引物的序列为UXS1ID1R:5'-GTAACCACGATACGCAAACTC-3';优选的在本发明中针对InDel1设计的正向引物SEQ ID NO.1的5'末端采用荧光基团FAM进行修饰。
在本发明中,由于InDel2和InDel3在尿苷二磷酸葡糖醛酸脱羧酶基因1的位点相距较近,因此,设计同一对引物对上述两个位点同时进行扩增;优选的根据InDel2和InDel3设计的引物为正向引物SEQ ID NO.3和反向引物SEQ ID NO.4;正向引物的序列为UXS1ID2F:5'-GTGGTTTTACTCGGTGCTTCT-3';反向引物的序列为UXS1ID2R:5'-GGCTCCAACTCAGTTCACACTT-3';优选的在本发明中针对InDel1设计的正向引物SEQ ID NO.3的5'末端采用荧光基团FAM进行修饰。
本发明中,根据InDel4设计的引物为正向引物SEQ ID NO.5和反向引物SEQ ID NO.6;正向引物的序列为UXS1ID4F:5'-TTCAGCACTCTACCAGTCCA-3';反向引物的序列为UXS1ID4R:5'-TCAGGCACACCACGTCAAA-3';优选的在本发明中针对InDel4设计的正向引物SEQ ID NO.5的5'末端采用荧光基团FAM进行修饰。
在本发明中,根据InDel5设计的引物为正向引物SEQ ID NO.7和反向引物SEQ ID NO.8;正向引物的序列为UXS1ID5F:5'-GGCTCTCTTCCGTAATCCA-3';反向引物的序列为UXS1ID5R:5'-CATTGCAAGGGAAAAACTACAC-3';优选的在本发明中针对InDel5设计的正向引物SEQ ID NO.7的5'末端采用荧光基团HEX进行修饰。
得到PCR扩增引物后,本发明用所述PCR扩增引物对待测样本全基因组DNA进行PCR扩增获得扩增产物,优选的,所述PCR扩增的条件为:95℃预变性5min;95℃变性30s,53~57℃退火30s,72℃延伸30s,35个循环;最后72℃延伸5min。所述退火温度更优选的为55~56℃;所述PCR扩增的体系优选的包括以下组分:0.7μl的待测样本全基因组DNA,0.15μl的0.8UTaq酶,0.3μl 0.2mM的dNTPs,0.9μl 25mM的MgCl2,1.25μl的10×PCR buffer,0.4μl 100nmolL-1的正向引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。
在本发明中所述待测样本全基因组DNA采用本领域常规的基因组DNA的提取方法获得,具体在本发明实施例中采用DNA提取试剂盒获得,具体的可采用sigma公司的植物基因组DNA小量制备试剂盒;所述基因组DNA的浓度优选的为5~20ng/μl,更优选的为10~15ng/μl。
本发明在获得扩增产物后,用毛细管荧光凝胶电泳检测扩增产物的片段大小,本发明对所述的毛细管荧光凝胶电泳的方法和具体参数设置没有特殊要求,采用本领域常规的方法和参数即可。所述毛细管荧光凝胶电泳检测的原理如下:当扩增片段经过扫描仪CCD(charge-coupled device)扫描窗口时,荧光染料分子发出一定波长的荧光被CCD接收,CCD记录每一个DNA片段通过的时间并以荧光峰值表示。理论上,荧光峰值出现的时间与扩增产物片段大小呈正相关,而峰值的高低与扩增产物的数量呈正相关。因此,在正向引物5'末端进行荧光基团修饰后,进行PCR扩增,通过检测扩增产物荧光片段大小的差异,可以判断目标区域靶位点的InDel类型。在本发明中,具体的,若扩增产物中有两条条带则该InDel位点的基因型为杂合型ID;若有一条条带且片段大小与杂合型ID中大的片段相同,则该InDel位点基因型为插入纯合型II;若有一条条带且片段大小与杂合型ID中小的片段相同,则表示该InDel位点为缺失纯合型DD。
下面结合实施例对本发明提供的InDel位点基因型分型的方法进行详细的说明,但是不能把它们理解为对本发明保护范围的限定。
实施例1
材料取自山东冠县毛白杨基因库(全国毛白杨种质资源库)中435株个体。
提取每株个体的DNA后,按照地域分布,随机挑选45株个体,逐一对其尿苷二磷酸葡糖醛酸脱羧酶基因1进行克隆测序,利用MEGA软件中的ClustalW程序进行多重比对,确定候选InDel1位点。InDel1位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第401bp到409bp处,其所在区域的基因序列如:斜体为InDel1位点。
针对InDel1位点(黑体下划线标注),整个自然群体包含两种类型的DNA模板链,其中模板链I:模板链II:3'-GAGGGCGGAATTGGGTAGACTAGGTTGGTGGGTAAGAAGAGAGGGGAGAAGTTAAATGGTA---------NNNNNNNNNNNN-5'。设计合成一条5'末端用FAM荧光基团进行修饰的正向引物:SEQ ID NO.1为UXS1ID1F:5'-CTCCCGCCTTAACCCATCT-3',一条不进行荧光修饰的反向引物:SEQ ID NO.2为UXS1ID1R:5'-GTAACCACGATACGCAAACTC-3',扩增产物目的片段长度为168bp。
将已知InDel基因型为DD、ID和II的三个待测样品的基因组DNA模板与所设计引物组合,进行PCR扩增,扩增程序为:预变性95℃5min,变性95℃30s,退火56℃30s,延伸72℃30s,35个循环,最后延伸72℃5min,4℃保存。12.5μl反应体系中包括:0.7μl 15ng/μl基因组DNA,0.15μl 0.8UTaq酶,0.3μl 0.2mM dNTPs,0.9μl 25mmolL-1MgCl2,1.25μl 10×PCRbuffer以及0.4μl100nmolL-1正向荧光标记引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。将PCR扩增的产物进行毛细管荧光凝胶电泳检测,每条泳道检测一株个体的基因型,根据检测结果确定待测样品的基因型。结果如图1所示,图1中上方为毛细管荧光电泳成像图,下部为荧光峰值图。其中,三个泳道内,字母DD表示该个体InDel1位点的基因型为deletion/deletion;字母ID表示该个体InDel1位点的基因型为insertion/deletion;字母II表示该个体InDel1位点的基因型为insertion/insertion。深灰色亮带为核苷酸分子量Marker,最右侧数值为核苷酸分子量大小。根据目的片段的大小,可以确定三株个体的基因型分别是DD、ID和II。
然后再取自山东冠县毛白杨基因库中435株个体中按照上述方法进行InDel1位点的基因型检测,其中有27株个体基因型为DD,有383株个体基因型为ID,有25株个体基因型为II。
实施例2
材料取自山东冠县毛白杨基因库(全国毛白杨种质资源库)中435株个体。
提取每株个体的DNA后,按照地域分布,随机挑选45株个体,逐一对其尿苷二磷酸葡糖醛酸脱羧酶基因1进行克隆测序,利用MEGA软件中的ClustalW程序进行多重比对,确定候选InDel2和InDel3位点。InDel2位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第1399bp到1401bp处,InDel3位点位于第1427bp到1438bp处,其所在区域的基因序列如下:斜体为InDel2位点;斜体为InDel3位点)进行基因型检测。
设计合成一条5'末端用FAM荧光基团进行修饰的正向引物:SEQ ID NO.3为UXS1ID2F:5'-GTGGTTTTACTCGGTGCTTCT-3',一条不进行荧光修饰的反向引物:SEQ ID NO.4为UXS1ID2R:5'-GGCTCCAACTCAGTTCACACTT-3',扩增产物目的片段长度为209bp。
按照实施例1的PCR扩增程序和体系对山东冠县毛白杨基因库中435株个体尿苷二磷酸葡糖醛酸脱羧酶基因1中的InDel2和InDel3位点基因型进行检测,检测结果为:有373株个体基因型为ID,有62株个体基因型为II;InDel3位点基因型检测结果为:435株个体基因型均为ID。
实施例3
材料取自山东冠县毛白杨基因库(全国毛白杨种质资源库)中435株个体。
提取每株个体的DNA后,按照地域分布,随机挑选45株个体,逐一对其尿苷二磷酸葡糖醛酸脱羧酶基因1进行克隆测序,利用MEGA软件中的ClustalW程序进行多重比对,确定候选InDel4和InDel5位点。InDel4位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第2094bp到2103bp处,其所在区域基因序列如下:斜体为InDel4位点;InDel5位于尿苷二磷酸葡糖醛酸脱羧酶基因1全长第4143bp到4152bp处,其所在区域的基因序列如下:斜体为InDel5位点,同时进行多位点基因型检测。
根据待检测位点InDel4和DNA模板链的序列特点,设计合成一条5'末端用FAM荧光基团进行修饰的正向引物,SEQ ID NO.5为UXS1ID4F:5'-TTCAGCACTCTACCAGTCCA-3'和一条不进行荧光修饰的反向引物SEQ ID NO.6为UXS1ID4R:5'-TCAGGCACACCACGTCAAA-3',扩增产物目的片段长度为160bp。
根据待检测位点InDel5和DNA模板链的序列特点,设计合成一条5'末端用HEX荧光基团进行修饰的正向引物:SEQ ID NO.7为UXS1ID5F:5'-GGCTCTCTTCCGTAATCCA-3'和一条不进行荧光修饰的反向引物SEQ ID NO.8为UXS1ID5R:5'-CATTGCAAGGGAAAAACTACAC-3',扩增产物目的片段长度为125bp。
将待测样品的基因组DNA模板与上述SEQ ID NO.5~SEQ ID NO.7引物组合,进行PCR扩增,扩增程序为:预变性95℃5min,变性95℃30s,退火57℃30s,延伸72℃30s,35个循环,最后延伸72℃5min,4℃保存。12.5μl反应体系中包括:0.7μl 15ng/μl基因组DNA,0.15μl 0.8UTaq酶,0.3μl 0.2mM dNTPs,0.9μl 25mmolL-1MgCl2,1.25μl 10×PCR buffer以及0.4μl 100nmolL-1正向荧光标记引物和0.4μl 100nmolL-1反向引物和8.4μl的ddH2O。将PCR扩增的产物进行毛细管荧光凝胶电泳检测,每条泳道检测一株个体的基因型,根据检测结果确定待测样品的基因型。对山东冠县毛白杨基因库中435株个体尿苷二磷酸葡糖醛酸脱羧酶基因1中的InDel4和InDel5位点基因型进行检测,InDel4位点检测结果为:有22株个体基因型为DD,有413株个体基因型为ID。InDel5位点基因型检测结果为:有364株个体基因型为ID,有71株个体基因型为II。
由以上实施例可知,应用本发明所述的InDel位点基因型分型方法,对引物组中的正向引物5'末端采用荧光基团进行修饰,在最佳退火温度和PCR反应条件下,可以显著地保留荧光信号强度;利用毛细管荧光电泳,通过检测荧光峰值出现的时间和峰值来判断扩增产物片段大小和数量,进而判断靶位点的InDel类型,实现对群体中所有个体的待测InDel位点进行基因型分型。应用毛细管荧光凝胶电泳技术检测扩增产物大小,将检测精度提高到1bp以内;同时,通过在正向引物5'末端修饰不同的荧光基团,可同时对多位点的基因型进行分型。因此不仅增加了检测结果的精确性和可靠性,而且极大地简化了实验步骤和配套条件,并显著地降低了实验成本。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!