一种甲哌鎓原药的合成方法与流程
本发明涉及药物合成工艺技术领域,具体涉及一种甲哌鎓原药的合成方法。
二、
背景技术:
:
甲哌鎓为新型植物生长调节剂,对植物有较好的内吸传导作用;甲哌鎓能够促进植物的生殖生长、抑制茎叶疯长、控制侧枝、塑造理想株型、提高根系数量和活力;并能使果实增重、品质提高。甲哌鎓广泛应用于棉花、小麦、水稻、花生、玉米、马铃薯、葡萄、蔬菜、豆类和花卉等农作物。甲哌鎓是一种高效、低毒、无药害内吸性药剂。根据用量和植物不同生长期喷洒甲哌鎓,可调节植物生长、使植株坚实抗倒伏、改进色泽和增加产量。棉花使用甲哌鎓能促进根系发育、叶色发绿、变厚防止徒长、抗倒伏、提高成铃率和增加霜前花,并使棉花品级提高,同时使株型紧凑、赘芽大大减少,节省整枝用工。甲哌鎓可被根、嫩枝、叶片吸收,很快传导到其它部位,不残留、不致癌。由于我国是农业大国,所以每年都需要大量的甲哌鎓。
目前,甲哌鎓的合成工艺主要是两步法:第一步是合成N-甲基哌啶,利用六氢哌啶与氯甲烷一步反应,除去HCl后得到N-甲基哌啶(见式1);N-甲基哌啶在加压条件下与氯甲烷反应生成甲哌鎓(见式2)。该工艺非常成熟,由于第一步需要把N-甲基哌啶分离出来才能进行下一步反应,存在耗时长、耗能大等问题。
为了简化工艺,现有文献中也出现过一种一步法合成甲哌鎓的方法,即哌啶与氯甲烷在过量缚酸剂K2CO3作用下,一步合成得到高纯度甲哌鎓,同时体系中得到的KHCO3和能够通过煅烧循环利用,有效节约成本,降低能耗(见式3)。
但是,目前所有合成甲哌鎓的方法都是以哌啶为原料,成本高并且不易储存。
三、
技术实现要素:
:
本发明要解决的技术问题是:针对现有甲哌鎓合成方法存在的技术问题,本发明提供一种利用吡啶为原料合成高纯度甲哌鎓的方法。
为了解决上述问题,本发明采取的技术方案是:
本发明提供一种甲哌鎓原药的合成方法,所述合成方法包括以下步骤:
a、首先将乙醇加入反应容器中,然后加入原料吡啶进行混合,溶解后加入氯甲烷;接着加热升温至60~100℃,控制反应时的压力为0.3~1.0MPa,在此温度、压力条件下反应3~6h,反应结束后得到N-甲基吡啶氯化物;
所述吡啶与乙醇之间加入量的质量比为1:2.5~5;所述吡啶与氯甲烷之间加入量的摩尔比为1:1.1~1.6;
b、将步骤a得到的N-甲基吡啶氯化物和催化剂加入反应容器中,所述催化剂的加入量占N-甲基吡啶氯化物质量的3~5%;然后向反应器内通入氢气,加热升温至120~150℃,控制反应时的压力为5~7MPa,在此温度、压力条件下反应7~15h,反应结束后得到N-甲基哌啶盐酸盐体系;
所述N-甲基吡啶氯化物与氢气之间加入量的摩尔比为1:3.0~5.5;
c、在步骤b得到的N-甲基哌啶盐酸盐体系中加入缚酸剂进行中和反应,所述N-甲基哌啶盐酸盐体系与缚酸剂之间加入的摩尔比为1:1.05~1.11;中和反应后进行抽滤,过滤后得到N-甲基哌啶;
d、将步骤c得到的N-甲基哌啶溶于乙醇,得到N-甲基哌啶乙醇溶液,然后向反应体系内加入氯甲烷,加热升温至25~100℃,控制反应压力为0.1~0.4MPa,在此温度、压力条件下反应时间为3~6h;
所述N-甲基哌啶与乙醇之间加入量的质量比为1:2.5~5;所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.1~1.6;
e、将步骤d得到的甲哌鎓体系进行蒸发,蒸除乙醇和氯甲烷,得到高纯度的甲哌鎓。
根据上述的甲哌鎓原药的合成方法,步骤a中所述乙醇的质量百分浓度为95~99.5%。
根据上述的甲哌鎓原药的合成方法,步骤a中所述吡啶与氯甲烷之间的摩尔比为1:1.1~1.2。
根据上述的甲哌鎓原药的合成方法,步骤b中所述催化剂为双金属负载型催化剂Ru-Pd/C或PTP-20吡啶加氢催化剂。
根据上述的甲哌鎓原药的合成方法,步骤c中所述缚酸剂为乙醇钠、NaOH、Na2CO3或NaHCO3。
根据上述的甲哌鎓原药的合成方法,步骤d中所述乙醇的质量百分浓度为95~99.5%。
根据上述的甲哌鎓原药的合成方法,步骤d中所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.1~1.2。
根据上述的甲哌鎓原药的合成方法,步骤e中所述高纯度甲哌鎓的纯度为99.5%。
本发明的积极有益效果:
1、本发明采用的原料吡啶和氯甲烷,以及生成的甲哌鎓都溶于乙醇,可以有效提纯甲哌鎓原药的纯度;本发明工艺操作简单,原材料成本低廉、工艺废水少,经济成本低,所得产品甲哌鎓纯度高,适合大规模连续化、工业化生产,是一种合成高纯度植物生长调节剂甲哌鎓的新工艺。
2、本发明方法利用的原料吡啶、氯甲烷以及生成的甲哌鎓都溶于乙醇,同时缚酸剂使用量大大减少,从而可以有效提高甲哌鎓原药的纯度。利用本发明技术方案制备得到的甲哌鎓纯度达到99.5%。
3、本发明工艺生产过程中减少了缚酸剂的使用量,节约了生产用水成本;同时连续化特点强,有效节约能耗,是一种新型的生产工艺。因此,本发明具有显著的经济效益和社会效益。
四、附图说明:
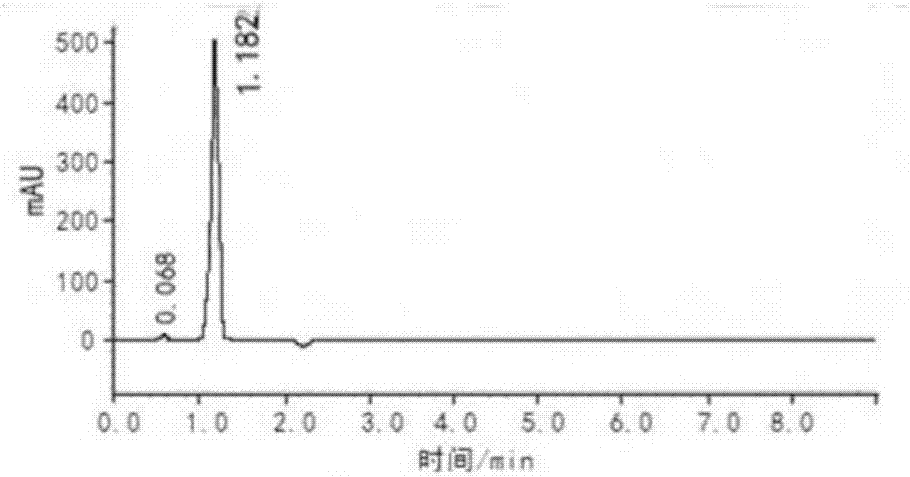
图1甲哌鎓标准样品的液相色谱图;
图2本发明实施例1所得产物的液相色谱图;
图3本发明实施例2所得产物的液相色谱图;
图4本发明实施例3所得产物的液相色谱图;
图5甲哌鎓标准样品工作曲线。
图6本发明实施例1所得甲哌鎓的核磁共振(1H-NMR)谱图。
五、具体实施方式:
以下结合实施例进一步阐述本发明,但并不限制本发明的保护内容。
实施例1:
本发明甲哌鎓原药的合成方法,该合成方法的详细步骤如下:
a、首先将质量百分浓度为99.5%的乙醇加入反应容器中,然后加入原料吡啶进行混合,溶解后加入氯甲烷;接着加热升温至80℃,控制反应时的压力为0.5MPa,在此温度、压力条件下反应6h,反应结束后得到N-甲基吡啶氯化物;
所述吡啶与乙醇之间加入量的质量比为1:2.5;所述吡啶与氯甲烷之间加入量的摩尔比为1:1.2;
b、将步骤a得到的N-甲基吡啶氯化物和双金属负载型催化剂Ru-Pd/C加入反应容器中,所述催化剂的加入量占N-甲基吡啶氯化物质量的4%;然后向反应器内通入氢气,所述N-甲基吡啶氯化物与氢气之间加入量的摩尔比为1:3.0;接着加热升温至130℃,控制反应时的压力为6MPa,在此温度、压力条件下反应12h,反应结束后得到N-甲基哌啶盐酸盐体系;
c、在步骤b得到的N-甲基哌啶盐酸盐体系中加入缚酸剂NaOH进行中和反应,所述N-甲基哌啶盐酸盐体系与缚酸剂NaOH之间加入的摩尔比为1:1.08;中和反应后进行抽滤,过滤后得到N-甲基哌啶;
d、将步骤c得到的N-甲基哌啶溶于质量百分浓度为99.5%的乙醇中,得到N-甲基哌啶乙醇溶液,然后向反应体系内加入氯甲烷,加热升温至60℃,控制反应压力为0.3MPa,在此温度、压力条件下反应时间为5h;
所述N-甲基哌啶与乙醇之间加入量的质量比为1:2.5;所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.2;
e、将步骤d得到的甲哌鎓体系进行蒸发,蒸除乙醇和氯甲烷,得到纯度为99.5%的甲哌鎓(详见附图2的液相色谱图)。
实施例2:
本发明甲哌鎓原药的合成方法,该合成方法的详细步骤如下:
a、首先将质量百分浓度为97%的乙醇加入反应容器中,然后加入原料吡啶进行混合,溶解后加入氯甲烷;接着加热升温至100℃,控制反应时的压力为0.7MPa,在此温度、压力条件下反应4h,反应结束后得到N-甲基吡啶氯化物;
所述吡啶与乙醇之间加入量的质量比为1:3.5;所述吡啶与氯甲烷之间加入量的摩尔比为1:1.1;
b、将步骤a得到的N-甲基吡啶氯化物和PTP-20吡啶加氢催化剂加入反应容器中,所述催化剂的加入量占N-甲基吡啶氯化物质量的3%;然后向反应器内通入氢气,所述N-甲基吡啶氯化物与氢气之间加入量的摩尔比为1:3.5;接着加热升温至150℃,控制反应时的压力为5MPa,在此温度、压力条件下反应8h,反应结束后得到N-甲基哌啶盐酸盐体系;
c、在步骤b得到的N-甲基哌啶盐酸盐体系中加入缚酸剂NaOH进行中和反应,所述N-甲基哌啶盐酸盐体系与缚酸剂NaOH之间加入的摩尔比为1:1.11;中和反应后进行抽滤,过滤后得到N-甲基哌啶;
d、将步骤c得到的N-甲基哌啶溶于质量百分浓度为97%的乙醇中,得到N-甲基哌啶乙醇溶液,然后向反应体系内加入氯甲烷,加热升温至100℃,控制反应压力为0.2MPa,在此温度、压力条件下反应时间为4.5h;
所述N-甲基哌啶与乙醇之间加入量的质量比为1:3.5;所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.1;
e、将步骤d得到的甲哌鎓体系进行蒸发,蒸除乙醇和氯甲烷,得到纯度为99.5%的甲哌鎓(详见附图3的液相色谱图)。
实施例3:
本发明甲哌鎓原药的合成方法,该合成方法的详细步骤如下:
a、首先将质量百分浓度为95%的乙醇加入反应容器中,然后加入原料吡啶进行混合,溶解后加入氯甲烷;接着加热升温至60℃,控制反应时的压力为0.9MPa,在此温度、压力条件下反应6h,反应结束后得到N-甲基吡啶氯化物;
所述吡啶与乙醇之间加入量的质量比为1:5;所述吡啶与氯甲烷之间加入量的摩尔比为1:1.2;
b、将步骤a得到的N-甲基吡啶氯化物和双金属负载型催化剂Ru-Pd/C加入反应容器中,所述催化剂的加入量占N-甲基吡啶氯化物质量的5%;然后向反应器内通入氢气,所述N-甲基吡啶氯化物与氢气之间加入量的摩尔比为1:3.0;接着加热升温至120℃,控制反应时的压力为7MPa,在此温度、压力条件下反应14h,反应结束后得到N-甲基哌啶盐酸盐体系;
c、在步骤b得到的N-甲基哌啶盐酸盐体系中加入缚酸剂NaOH进行中和反应,所述N-甲基哌啶盐酸盐体系与缚酸剂NaOH之间加入的摩尔比为1:1.05;中和反应后进行抽滤,过滤后得到N-甲基哌啶;
d、将步骤c得到的N-甲基哌啶溶于质量百分浓度为95%的乙醇中,得到N-甲基哌啶乙醇溶液,然后向反应体系内加入氯甲烷,加热升温至50℃,控制反应压力为0.35MPa,在此温度、压力条件下反应时间为6h;
所述N-甲基哌啶与乙醇之间加入量的质量比为1:5;所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.2;
e、将步骤d得到的甲哌鎓体系进行蒸发,蒸除乙醇和氯甲烷,得到纯度为99.5%的甲哌鎓(详见附图4的液相色谱图)。
实施例4:
本发明甲哌鎓原药的合成方法,该合成方法的详细步骤如下:
a、首先将质量百分浓度为98%的乙醇加入反应容器中,然后加入原料吡啶进行混合,溶解后加入氯甲烷;接着加热升温至70℃,控制反应时的压力为1.0MPa,在此温度、压力条件下反应5h,反应结束后得到N-甲基吡啶氯化物;
所述吡啶与乙醇之间加入量的质量比为1:2.8;所述吡啶与氯甲烷之间加入量的摩尔比为1:1.3;
b、将步骤a得到的N-甲基吡啶氯化物和PTP-20吡啶加氢催化剂加入反应容器中,所述催化剂的加入量占N-甲基吡啶氯化物质量的4.5%;然后向反应器内通入氢气,所述N-甲基吡啶氯化物与氢气之间加入量的摩尔比为1:3.0;接着加热升温至140℃,控制反应时的压力为6MPa,在此温度、压力条件下反应12h,反应结束后得到N-甲基哌啶盐酸盐体系;
c、在步骤b得到的N-甲基哌啶盐酸盐体系中加入缚酸剂乙醇钠进行中和反应,所述N-甲基哌啶盐酸盐体系与缚酸剂乙醇钠之间加入的摩尔比为1:1.08;中和反应后进行抽滤,过滤后得到N-甲基哌啶;
d、将步骤c得到的N-甲基哌啶溶于质量百分浓度为98%的乙醇中,得到N-甲基哌啶乙醇溶液,然后向反应体系内加入氯甲烷,加热升温至70℃,控制反应压力为0.3MPa,在此温度、压力条件下反应时间为5h;
所述N-甲基哌啶与乙醇之间加入量的质量比为1:3.0;所述N-甲基哌啶与氯甲烷之间的摩尔比为1:1.3;
e、将步骤d得到的甲哌鎓体系进行蒸发,蒸除乙醇和氯甲烷,得到纯度为99.5%的甲哌鎓。
另外,对本发明实施例1所得产品甲哌鎓进行了核磁共振谱图分析即(1H-NMR)谱图,详见附图6。
- 还没有人留言评论。精彩留言会获得点赞!