一种基于稠环呋喃的小分子材料制备方法及其应用与流程
本发明属于分子材料技术领域,尤其涉及一种基于稠环呋喃的小分子材料制备方法及其应用。
背景技术:
有机半导体器件主要包括有机发光二极管(OLEDs)、有机场效应晶体管(OFETs)以及有机太阳能电池(OPVs)等。目前,OLEDs已经在一些小型设备,如移动电话、掌上电脑、数码相机以及路灯等,实现了商业化应用。而OFETs和OPVs在实用化方面取得了很大的进展,正在向产业化迈进。提升有机半导体器件性质的重要途径之一就是开发新型有机分子材料。有机分子材料是一类含有π-电子结构、具有光、电、磁性质的有机光电子材料,也称有机半导体材料。相比于无机材料,有机半导体材料具有易于调控,制备工艺简单,成本低廉,易于大面积制备有机柔性电路等特点。近几十年来,有机分子材料的研发引起了广泛的关注,从而促使有机半导体器件性质不断提升。比如,并五苯的单晶晶体管在室温下的迁移率高达35 cm2V-1s-1。稠环呋喃化合物是一类重要的共轭分子材料,在有机半导体器件领域有着非常广泛的应用,例如,基于2,7-二(4-辛基苯基)萘[2,1-b:6,5-b′]二呋喃的OFETs器件空穴迁移率可达3.6 cm2V-1s-1。通过呋喃成环反应来合成稠环呋喃小分子材料,常见的三种方法如下:
(1)方法一:Pd催化的分子内乌尔曼 C-O偶联反应
该方法产率高,但是底物合成复杂,需要使用昂贵的钯催化剂和膦配体,且反应温度较高(100 oC),反应时间长(≥24 h),因此实用价值较低 [参见K. Kawaguchi, K. Nakano, K. Nozaki, J. Org. Chem. 2007, 72, 5119]。
(2)方法二:Pd催化的二芳基醚的双C-H活化关环反应
该方法产率较高,但是底物合成复杂,需要使用昂贵的钯催化剂和银氧化剂。且反应温度较高(≥110 oC),反应时间长(≥ 20 h),因此实用价值较低。
(3)方法三:邻羟基芳基炔的关环反应
该方法产率高,但是需要使用等当量的对水敏感的正丁基锂、无水氯化锌,还需要使用无水溶剂,且反应温度较高(120 oC)。另外正丁基锂特别危险,因此实用价值较低。
综上所述,现有合成稠环呋喃小分子材料的技术存在的问题是:1)底物合成复杂,耗时费力;2)方法一和二使用昂贵的钯催化剂,成本较高,而方法三需要使用对水敏感的正丁基锂,不方便操作;3)由于呋喃成环反应需要克服较大的能垒,所以需要较高的反应温度(≥100 oC)。因此,以上技术的实用价值较低。
技术实现要素:
针对现有技术存在的问题,本发明提供了一种基于稠环呋喃的小分子材料制备新方法及其应用。
本发明是这样实现的,一种基于稠环呋喃小分子材料的制备方法,所述基于稠环呋喃小分子材料的制备方法包括以下步骤:
步骤一,以 2,3-二溴呋喃、2,3-二溴噻吩、2,3-二溴硒吩、2,3-二溴-吲哚衍生物和2-羟基苯硼酸衍生物为原料,经过铃木反应得到关键中间体;
步骤二,以亚铜或铜化合物为催化剂,1,10-菲啰啉或联吡啶化合物为配体,发生分子内乌尔曼 C-O偶联反应,合成了一系列稠环呋喃类小分子材料。
进一步,所述基于稠环呋喃小分子材料的制备方法合成路线如下:
进一步,所述 Ar, Ar'选自芳基、杂芳基、含有取代基的芳基和含有取代基的杂芳基中的任意一种;取代基R, R'为氢、卤素、C1-C30烷基、C1-C30烷氧基、芳基、酰基、酯基、硝基、氨基、(烷基)胺基中的一种或多种,取代基的位置为Ar, Ar'环上的任意位置;N上取代基R''为氢、C1-C30烷基、芳基中的一种或多种;铜催化剂是碘化亚铜、溴化亚铜、氯化亚铜、氧化亚铜、噻吩-2-甲酸亚铜、氧化铜、氯化铜中任意一种;碱是碳酸钾、碳酸钙、碳酸铯、碳酸钠、碳酸氢钠、乙酸钠、乙酸钾、乙醇钠、叔丁醇钠、叔丁醇钾、氢氧化锂、氢氧化钠、磷酸钾中任意一种;配体是1,10-菲啰啉、2,9-二甲基-1,10-菲啰啉、2,2'-联吡啶、4,4'-二甲基-2,2'-联吡啶、4,4'-二壬基-2,2'-联吡啶或4,4'-二叔丁基-2,2'-联吡啶中任意一种。
进一步,四(三苯基膦)钯催化剂,铜催化剂以及配体的加入量一般为反应起始物的0.05-0.1当量。
进一步,铃木反应条件为:化合物1(1当量),化合物2(1.5当量)加入到1,4-二氧六环和水的混合溶剂中(体积比4:1),碳酸钾(4当量)被加入到该混合溶剂中,通氮气除氧,四(三苯基膦)钯(0.05当量)加入到该体系中,90 ℃反应6小时。
进一步,所述铜催化剂催化乌尔曼分子内C-O偶联的反应条件为:化合物3(1 当量)加入到溶剂N,N-二甲基甲酰胺中,通氮气除氧,碳酸钾(4当量)被加入到该混合溶剂中,铜催化剂(0.05当量)以及配体(0.1当量)加入到该体系中,90 ℃反应15分钟。
本发明的另一目的在于提供一种所述基于稠环呋喃小分子材料的制备方法制备的基于稠环呋喃小分子材料,所述基于稠环呋喃小分子材料的分子结构如下:
本发明的另一目的在于提供一种由所述基于稠环呋喃小分子材料制造的有机半导体器件。
本发明的另一目的在于提供一种由所述有机半导体器件制造的移动电话。
本发明的另一目的在于提供一种由所述有机半导体器件制造的掌上电脑。
本发明的优点及积极效果为:本发明底物的合成相对简单,产率高;呋喃成环反应时间大大缩短(15 min);铜催化剂和联吡啶化合物为催化体系比较便宜;反应条件更温和(90℃);底物适用范围广;反应产率高,最高可达97%。因此该方法适合于工业化生产。
附图说明
图1是本发明实施例提供的基于稠环呋喃小分子材料的制备方法流程图。
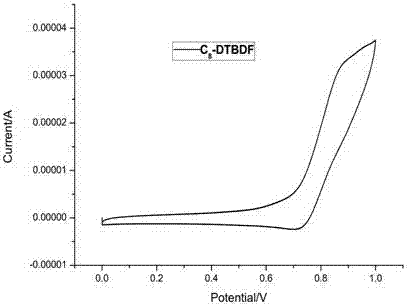
图2是本发明实施例提供的C8-DTBDF的循环伏安曲线(溶剂二氯甲烷, 最高占据轨道能级: -5.42 eV)示意图。
图3是本发明实施例提供的FET转移特性曲线和输出特性曲线示意图。
具体实施方式
为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
下面结合附图对本发明的应用原理作详细的描述。
本发明实施例提供的基于稠环呋喃小分子材料分子结构如下:
如图1所示,本发明实施例提供的基于稠环呋喃小分子材料的制备方法包括以下步骤:
S101:以 2,3-二溴呋喃、2,3-二溴噻吩、2,3-二溴硒吩、2,3-二溴-吲哚衍生物和2-羟基苯硼酸衍生物为原料,经过铃木反应得到关键中间体3;
S102:以亚铜或铜化合物(铜催化剂)为催化剂,1,10-菲啰啉等联吡啶化合物为配体,3发生分子内乌尔曼 C-O偶联反应,合成了一系列稠环呋喃类小分子材料4。
本发明实施例提供的基于稠环呋喃小分子材料的制备方法合成路线如下:
其中Ar, Ar'选自芳基、杂芳基、含有取代基的芳基和含有取代基的杂芳基中的任意一种。取代基R, R'为氢、卤素、C1-C30烷基、C1-C30烷氧基、芳基、酰基、酯基、硝基、氨基、(烷基)胺基中的一种或多种,取代基的位置为Ar, Ar'环上的任意位置。铜催化剂是碘化亚铜、溴化亚铜、氯化亚铜、氧化亚铜、噻吩-2-甲酸亚铜、氧化铜、氯化铜中任意一种。碱是碳酸钾、碳酸钙、碳酸铯、碳酸钠、碳酸氢钠、乙酸钠、乙酸钾、乙醇钠、叔丁醇钠、叔丁醇钾、氢氧化锂、氢氧化钠、磷酸钾中任意一种。配体是1,10-菲啰啉、2,9-二甲基-1,10-菲啰啉、2,2'-联吡啶、4,4'-二甲基-2,2'-联吡啶、4,4'-二壬基-2,2'-联吡啶或4,4'-二叔丁基-2,2'-联吡啶中任意一种。
在上述合成方法的反应中,所用的四(三苯基膦)钯催化剂,铜催化剂以及配体的加入量一般为反应起始物的0.05-0.1当量。
铃木反应条件为:化合物1(1当量),化合物2(1.5当量)加入到1,4-二氧六环和水的混合溶剂中(体积比4:1),碳酸钾(4当量)被加入到该混合溶剂中,通氮气除氧,四(三苯基膦)钯(0.05当量)加入到该体系中,90 ℃反应6小时。
铜催化剂催化乌尔曼分子内C-O偶联的反应条件为:化合物3(1当量)加入到溶剂N,N-二甲基甲酰胺或N-甲基吡咯烷酮中,通氮气除氧,碳酸钾(4 当量)被加入到该混合溶剂中,铜催化剂(0.05当量)以及配体(0.1当量)加入到该体系中,90 ℃反应15分钟。
在上述合成方法的反应中,所用的四(三苯基膦)钯催化剂,铜催化剂以及配体的加入量一般为反应起始物的0.05-0.1当量。
铃木反应条件为:化合物1(1当量),化合物2(1.5当量)加入到1,4-二氧六环和水的混合溶剂中(体积比4:1),碳酸钾(4当量)被加入到该混合溶剂中,通氮气除氧,四(三苯基膦)钯(0.05当量)加入到该体系中,90 ℃反应6小时。
化合物3(1当量)加入到溶剂N,N-二甲基甲酰胺中,通氮气除氧,碳酸钾(4 当量)被加入到该混合溶剂中,碘化亚铜(0.05当量)以及配体(0.1当量)加入到该体系中,90 ℃反应15分钟。
通过铜催化的、快速高效的C-O偶联关环反应来合成呋喃类化合物,包括稠环呋喃并呋喃、稠环噻吩并呋喃、稠环硒吩并呋喃以及稠环吲哚并呋喃及其衍生物(烷基链取代、芳香化合物取代、卤素取代以及羰基、硝基等取代基团);
铜催化剂为碘化亚铜、溴化亚铜、氯化亚铜、氧化亚铜、噻吩-2-甲酸亚铜等一价铜,以及氧化铜、氯化铜等二价铜;
配体包括1,10-菲啰啉、2,9-二甲基-1,10-菲啰啉、2,2'-联吡啶、4,4'-二甲基-2,2'-联吡啶、4,4'-二壬基-2,2'-联吡啶或4,4'-二叔丁基-2,2'-联吡啶等双齿配体;
铜催化剂催化乌尔曼分子内C-O偶联的溶剂为N,N-二甲基甲酰胺或甲苯;
铜催化剂催化乌尔曼分子内C-O偶联的反应温度一般为90 ℃;
碱为碳酸钾、碳酸钙、碳酸铯、碳酸钠、碳酸氢钠、乙酸钠、乙酸钾、乙醇钠、叔丁醇钠、叔丁醇钾、氢氧化锂、氢氧化钠、磷酸钾等,优先选择碳酸钾;
铜催化剂催化乌尔曼分子内C-O偶联的反应时间一般为15min;
铃木反应催化剂为四三苯基磷钯,溶剂为二氧六环和水(体积比4:1);
Pd催化选择性铃木反应;
基于本专利中的稠环呋喃类小分子材料的有机场效应晶体管的制备和应用。
下面结合具体实施例对本发明的应用原理作进一步的描述。
实施例1 苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]噻吩(683 mg, 2.34 mmol)、2-羟基苯硼酸(484 mg, 3.5 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (1.29 g, 9.36 mmol),四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴苯并[b]噻吩-2-基)苯酚 3a,产率为 84%。1H NMR (300 MHz, 氘代氯仿): δ 7.86-7.78 (m, 2H), 7.47-7.32 (m, 4H), 7.04-6.98 (m, 2H), 5.35 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 153.42, 138.82, 138.39, 133.55, 131.78, 131.29, 125.96, 125.52, 123.73, 122.45, 120.76, 119.09, 116.44, 108.63. 质谱(EI模式):m/z C14H9BrOS [M]+ 304。
2)步骤1)所得产物 (884 mg, 2.9 mmol),碳酸钾 (800 mg, 5.8 mmol),碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中, 90 oC反应15 min, 柱色谱分离得到目标化合物4a (520 mg, 2.32 mmol, 80% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 8.01 (d, J = 7.8 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.73 (d, J = 6.9 Hz, 1H), 7.65 (d, J = 7.8 Hz, 1H), 7.50-7.32 (m, 4H). 13C NMR (75 MHz, 氘代氯仿): δ 158.83, 153.04, 142.04, 125.18, 124.98, 124.95, 124.41, 124.10, 123.34, 119.74, 119.63, 118.62, 112.60。
实施例2 噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴噻吩(504 mg, 2.1 mmol)、2-羟基苯硼酸(433 mg, 3.1 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (828 mg,6 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴苯[b]噻吩-2-基)苯酚3b,产率为 87%。1H NMR (400 MHz, 氘代氯仿): δ 7.43 (d, J = 5.2 Hz, 1H), 7.39-7.32 (m, 1H), 7.28 (dd, J = 8.8, 1.2 Hz, 1H), 7.12 (d, J = 5.6 Hz, 1H), 7.01-6.98 (m, 2H), 5.13 (s, 1H). 13C NMR (101 MHz, 氘代氯仿): δ 153.42, 133.16, 131.88, 131.04, 130.96, 127.37, 120.63, 118.80, 116.21, 111.05. 高分辨质谱(EI模式):m/z for C10H7BrOS [M]+ 计算值253.9401, 实测值 253.9399。
2)步骤1)所得产物 (160 mg, 0.63 mmol),碳酸钾 (168 mg, 1.22 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4b (67 mg, 0.39 mmol, 产率 62%). 1H NMR (300 MHz, 氘代氯仿): δ 7.68-7.65 (m, 1H), 7.57-7.54 (m, 1H), 7.36 (d, J = 5.3 Hz, 1H), 7.34-7.28 (m, 2H), 7.15 (d, J = 5.3 Hz, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 159.35, 159.24, 127.56, 124.40, 123.82, 123.02, 119.39, 119.11, 112.49, 111.51。
实施例3 2-(叔丁基)苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴-6-叔丁基苯并[b]噻吩(810 mg, 2.3 mmol)、2-羟基苯硼酸(390 mg, 2.82 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (3.0 当量)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴-6-叔丁基苯并[b]噻吩-2-基)苯酚3c,产率为 88%。1H NMR (300 MHz, 氘代氯仿): δ 7.83 (s, 1H), 7.79 (d, J = 7.8Hz 1H), 7.56 (dd, J = 7.8, 1.5 Hz, 1H), 7.39-7.33 (m, 2H), 7.04-6.98 (m, 2H), 5.31 (s, 1H), 1.40 (s,9H). 13C NMR (75 MHz, 氘代氯仿) δ 153.44, 149.70, 138.98, 136.20, 132.52, 131.74, 131.20, 123.87, 123.21, 120.69, 119.19, 118.57, 116.35, 108.29, 35.17, 31.56. 质谱(EI模式):m/z 360。
2)步骤1)所得产物 (95 mg, 0.26 mmol),碳酸钾 (70 mg, 0.5 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4c (60 mg, 0.21 mmol, 81% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.91 (d, J = 8.5 Hz, 1H), 7.86 (d, J = 1.5 Hz, 1H), 7.71-7.65 (m, 1H), 7.65-7.58 (m, 1H), 7.51 (dd, J = 8.4, 1.7 Hz, 1H), 7.38-7.27 (m, 2H), 1.40 (s, 9H). 13C NMR (75 MHz, 氘代氯仿): δ 158.74, 153.05, 148.59, 142.46, 124.62, 124.32, 123.27, 123.23, 122.94, 120.68, 119.47, 119.26, 117.94, 112.53, 35.19, 31.57. 高分辨质谱(EI模式): m/z for C18H16OS [M]+ 计算值280.0922, 实测值 280.0923。
实施例4 7-(叔丁基)苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]噻吩(300 mg, 1.0 mmol)、4-叔丁基-2-羟基苯硼酸(300 mg, 1.5 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (3.0 当量)、四(三苯基膦)钯 (5% mol),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴-苯并[b]噻吩-2-基)-5-叔丁基苯酚3d,产率为 76%。1H NMR (400 MHz, 氘代氯仿): δ 7.85 (d, J= 8.0 Hz, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.50-7.39 (m, 2H), 7.30 (d, J = 8.0 Hz, 1H), 7.08-7.04 (m, 2H), 5.28 (s, 1H), 1.35 (s, 9H). 13C NMR (101 MHz, 氘代氯仿): δ 155.13, 152.98, 138.77, 138.45, 133.89, 131.21, 125.82, 125.44, 123.64, 122.40, 118.04, 116.01, 113.54, 108.23, 34.92, 31.24. 质谱(EI模式):m/z 360。
2)步骤1)所得产物 (83 mg, 0.23 mmol),碳酸钾 (70 mg, 0.5 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(0.1 M)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4d (56 mg, 0.2 mmol, 87% 产率)。该化合物的表征数据如下:1H NMR (400 MHz, 氘代氯仿): δ 7.98 (d, J = 8.0 Hz, 1H), 7.87 (d, J = 8.0 Hz, 1H), 7.67 (d, J = 1.2 Hz, 2H), 7.64 (d, J = 8.0 Hz), 7.8-7.43 (m, 3H), 1.42 (s, 9H). 13C NMR (101 MHz, 氘代氯仿): δ 159.33, 152.91, 149.16, 141.85, 125.36, 124.90, 124.63, 124.38, 121.49, 121.07, 119.53, 118.89, 118.57, 109.41, 35.19, 31.70. 高分辨质谱(EI模式):m/zfor C18H16OS [M]+ 计算值280.0922, 实测值 280.0922。
实施例5 2,7-二叔丁基苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴-6-叔丁基苯并[b]噻吩(238 mg, 0.66 mmol)、4-叔丁基-2-羟基苯硼酸(200 mg, 1.0 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (3.0 当量)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴-6-叔丁基苯并[b]噻吩-2-基)-5-叔丁基苯酚3e,产率为 63%。1H NMR (300 MHz, 氘代氯仿): δ 7.82 (d, J = 0.9 Hz , 1H), 7.79 (d, J = 8.4 Hz, 1H), 7.56 (s, J = 8.4, 1.5 Hz, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.05 (m, 2H), 5.25 (s, 1H), 1.40 (s, 9H), 1.35 (s, 9H). 13C NMR (75 MHz, 氘代氯仿): δ 155.02, 152.98, 149.56, 138.91, 136.25, 132.85, 131.18, 123.78, 123.12, 118.53, 117.97, 116.12, 113.45, 107.89, 35.13, 34.90, 31.54, 31.24. 高分辨质谱(EI模式):m/z for C22H25BrOS [M]+ 计算值416.0809, 实测值 416.0806。
2)步骤1)所得产物 (50 mg, 0.119 mmol),碳酸钾 (2当量) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4e (39 mg, 0.116 mmol, 97% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.91 (d, J = 8.4 Hz, 1H), 7.86 (s, 1H), 7.66 (s, 1H), 7.62 (d, J = 8.1 Hz, 1H), 7.51 (dd, J = 8.4, 1.5 Hz, 1H), 7.40 (dd, J = 8.4, 1.5 Hz, 1H), 1.41 (s, 18H). 13C NMR (75 MHz, 氘代氯仿): δ 159.17, 152.88, 148.79, 148.25, 142.21, 123.14, 123.08, 121.67, 120.98, 120.66, 119.04, 118.73, 117.83, 109.34, 35.16, 31.70, 31.57. 高分辨质谱 (MALDI-TOF模式):m/z for C22H24OS [M]+ 计算值336.1548, 实测值 336.1544。
实施例6 8-氟苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]噻吩(180 mg, 0.6 mmol)、5-氟-2-羟基苯硼酸(140 mg, 1.1 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (331mg,2.4 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴苯并[b]噻吩-2-基)-4-氟苯酚3f,产率为 63%。1H NMR (300 MHz, 氘代氯仿): δ 7.88-7.81 (m, 2H), 7.53-7.41 (m, 2H), 7.10-7.03 (m, 2H), 7.00 - 6.95 (m, 1H), 5.19 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 158.10, 154.93, 149.58 (d, J = 2.2 Hz), 138.73, 138.23, 132.23, 126.21, 125.65, 123.84, 122.47, 119.84 (d, J = 8.5 Hz), 118.09-117.47 (m), 108.99. 高分辨质谱(EI模式):m/z for C14H8BrFOS [M]+ 计算值321.9463,实测值 321.9462。
2)步骤1)所得产物 (60 mg, 0.19 mmol),碳酸钾 (52 mg, 0.38 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4f (42 mg, 0.17 mmol, 90% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ8.00 (d, J = 8.4 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.58-7.36 (m, 4H), 7.12-7.05 (m, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 160.89, 157.72, 154.93, 154.64, 142.21, 125.39, 125.10, 124.43, 119.91, 113.18 (d, J = 9.7 Hz), 112.52, 112.17, 105.60 (d, J = 26.2 Hz). 高分辨质谱 (MALDI-TOF模式):m/z for C14H7FOS [M]+ 计算值242.0196, 实测值 242.0197。
实施例7 1-{苯并[4,5]噻吩并[3,2-b]苯并呋喃}-辛-1-酮的合成
1)准确称取1-{2,3-二溴苯并[b]噻吩}-辛-1-酮的合成(210 mg, 0.5 mmol)、2-羟基苯硼酸(110 mg, 0.8 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (207mg,1.5 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到1-(3-溴-2-(2-羟基苯基)苯并[b]噻吩-6-基)辛-1-酮3g,产率为 66%。1H NMR (300 MHz, 氘代氯仿): δ 8.44 (s, 1H), 8.07 (dd, J = 8.4, 1.2 Hz, 1H), 7.92 (d, J = 8.4 Hz, 1H), 7.43-7.37 (m, 2H), 7.08-7.04 (m, 2H), 5.34 (s, 1H), 3.05 (t, J = 7.5 Hz, 2H), 1.77 (m, 2H), 1.31 (m, 8H), 0.89 (t, J = 6.9 Hz, 3H). 13C NMR (75 MHz, 氘代氯仿): δ 199.84, 153.28, 141.47, 138.73, 138.14, 134.44, 131.64, 131.56, 124.94, 123.68, 122.93, 120.84, 118.79, 116.59, 108.45, 38.98, 31.73, 29.36, 29.16, 24.53, 22.64, 14.10. 高分辨质谱 (MALDI-TOF模式):m/z for C22H23BrO2S [M+H]+ 计算值431.06804,实测值 431.06763。
2)步骤1)所得产物 (33 mg, 0.077 mmol),碳酸钾 (21 mg, 0.15 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4g (20 mg, 0.057 mmol, 74% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ8.52 (s, 1H), 8.10-8.03 (m, 2H), 7.78 (dd, J = 7.6, 1.1 Hz, 1H), 7.68 (d, J = 7.8 Hz, 1H), 7.47-7.36 (m, 2H), 3.06 (t, J = 7.5 Hz, 2H), 1.78 (m, 2H), 1.49-1.15 (m, 8H), 0.90 (t, J = 6.7 Hz, 3H). 13C NMR (75 MHz, 氘代氯仿): δ 199.58, 159.25, 152.35, 141.89, 133.52, 127.91, 125.98, 124.98, 124.84, 123.73, 123.64, 122.69, 120.10, 119.47, 112.81, 38.84, 31.75, 29.40, 29.18, 24.56, 22.66, 14.11. 高分辨质谱(EI模式):m/z for C22H22O2S [M]+ 350.1341, 实测值 350.1343。
实施例8 8-溴苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]噻吩(600 mg, 2.0 mmol)、5-溴-2-羟基苯硼酸(418 mg, 1.0 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (828 mg,6 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到4-溴-2-(3-溴苯并[b]噻吩-2-基)苯酚3h,产率为 78%。1H NMR (300 MHz, 氘代氯仿): δ 7.89-7.83 (m, 2H), 7.54-7.43 (m, 4H), 6.94 (dd, J = 8.4, 0.9 Hz, 1H), 5.25 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 152.61, 138.79, 138.19, 134.03, 133.94, 131.69, 126.27, 125.69, 123.85, 122.48, 120.96, 118.21, 112.51, 109.22. 高分辨质谱(EI模式):m/z for C14H8Br2OS [M]+ 计算值381.8662,实测值 381.8655。
2)步骤1)所得产物 (121 mg, 0.32 mmol),碳酸钾 (80 mg, 0.60 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4h (70 mg, 0.23 mmol, 72% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ8.01 (d, J = 7.5 Hz, 1H), 7.90-7.87 (m, 2H), 7.54-7.39 (m, 4H). 13C NMR (101 MHz, 氘代氯仿): δ 157.54, 154.07, 142.28, 127.70, 125.88, 125.45, 125.13, 124.82, 124.43, 122.33, 119.94, 117.66, 116.35, 113.93. 高分辨质谱 (MALDI-TOF模式):m/z for C14H7BrOS [M]+ 计算值301.9401, 实测值 301.9398。
实施例9 2-溴苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3,6-三溴苯并[b]噻吩(252 mg, 0.68 mmol)、2-羟基苯硼酸(93 mg, 0.67 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (281 mg,2.04 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3,6-二溴苯并[b]噻吩-2-基)苯酚3i,产率为 91%。1H NMR (300 MHz, 氘代氯仿): δ 7.98 (d, J = 1.5 Hz, 1H), 7.71 (d, J = 8.4 Hz, 1H), 7.59 (dd, J = 8.7, 1.8 Hz, 1H), 7.40-7.33 (m, 2H), 7.06-7.01 (m, 2H), 5.19 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 153.29, 140.07, 137.26, 134.25, 131.70, 131.45, 128.97, 124.88, 124.83, 120.85, 119.96, 118.64, 116.49, 108.25. 高分辨质谱(EI模式):m/z for C14H8Br2OS [M]+ 计算值383.8642, 实测值 383.8649。
2)步骤1)所得产物 (79 mg, 0.2 mmol),碳酸钾 (55 mg, 0.4 mmol)分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4i (40 mg, 66% 产率)。该化合物的表征数据如下:1H NMR (400 MHz, 氘代氯仿): δ 8.01 (s, 1H), 7.84 (d, J = 8.4 Hz, 1H), 7.71 (d, J = 7.6 Hz, 1H), 7.63 (d, J = 8.0 Hz, 1H), 7.57 (dd, J = 8.4, 1.2 Hz, 1H), 7.40-7.35 (m, 2H). 13C NMR (75 MHz, 氘代氯仿): δ158.89, 152.35, 143.29, 128.40, 126.89, 125.32, 123.93, 123.81, 123.54, 120.64, 119.68, 119.11, 118.46, 112.67. 高分辨质谱 (MALDI-TOF模式):m/z for C14H7BrOS [M]+ 计算值301.9401, 实测值 301.9397。
实施例10 8-氯苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]噻吩(300 mg, 1.0 mmol)、5-氯-2-羟基苯硼酸(300 mg, 1.6 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (414 mg,3 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴苯并[b]噻吩-2-基)-4-氯苯酚3j,产率为 82%。1H NMR (300 MHz, 氘代氯仿): δ 7.90-7.84 (m, 2H), 7.56-7.415 (m, 2H), 7.35- 7.32 (m, 2H), 7.00 (dd, J = 7.5, 1.8 Hz, 1H), 5.21 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 152.08, 138.77, 138.20, 131.79, 131.11, 131.06, 126.25, 125.67, 125.44, 123.84, 122.47, 120.39, 117.78, 109.16. 质谱(EI模式): m/z for C14H8BrClOS [M]+ 338。
2)步骤1)(16 mg, 0.047 mmol),碳酸氢钠 (16 mg, 0.2 mmol)分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4j (9 mg, 0.039 mmol, 74% 产率)。该化合物的表征数据如下:1H NMR (400 MHz, 氘代氯仿): δ 7.99 (d, J = 7.9 Hz, 1H), 7.88 (d, J = 8.1 Hz, 1H), 7.70 (d, J = 2.1 Hz, 1H), 7.56 (d, J = 8.8 Hz, 1H), 7.51-7.43 (m, 2H), 7.34 (dd, J = 8.8, 2.1 Hz, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 157.13, 154.23, 142.26, 128.96, 125.13, 124.97, 124.85, 124.43, 119.93, 119.32, 117.82, 113.47。
实施例11 3-氯苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴-5-氯苯并[b]噻吩(325 mg, 1.0 mmol)、2-羟基苯硼酸(220 mg, 1.6 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (552mg,4.0mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴-5-氯苯并[b]噻吩-2-基)苯酚3k,产率为 70%。1H NMR (400 MHz, 氘代氯仿) δ 7.84 (d, J = 2.0 Hz, 1H), 7.72 (d, J = 8.8 Hz, 1H), 7.40-7.33 (m, 3H), 7.05-7.01 (m, 2H), 5.19 (s, 1H). 13C NMR (101 MHz, 氘代氯仿) δ 153.25, 139.64, 136.89, 135.75, 132.00, 131.69, 131.47, 126.41, 123.49, 123.37, 120.86, 118.72, 116.51, 107.58. 质谱(EI模式):m/z C14H8BrClOS [M]+ 338。
2)步骤1)所得产物 (30 mg, 0.088 mmol),碳酸氢钠(30 mg, 0.35 mmol)分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 溴化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4k (14 mg, 0.054 mmol, 65% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ7.96 (d, J = 2.0 Hz, 1H), 7.78-7.70 (m, 2H), 7.63 (d, J = 8.1 Hz, 1H), 7.42-7.31 (m, 3H). 13C NMR (75 MHz, 氘代氯仿): δ 158.91, 151.84, 139.94, 131.35, 126.13, 125,48, 125.28, 125.19, 123.77, 123.51, 120.47, 119.78, 119.46, 112.73. 高分辨质谱(EI模式):m/z for C14H7ClOS [M]+ 计算值257.9906, 实测值 257.9903。
实施例12 3,8-二氯苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴-5-氯苯并[b]噻吩(330 mg, 1.0 mmol)、5-氯-2-羟基苯硼酸(220 mg, 1.5 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (552 mg,4.0 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴-5-氯苯并[b]噻吩-2-基)-4-氯苯酚3l,产率为 64%。1H NMR (300 MHz, 氘代氯仿): δ 7.87 (d, J = 2.1 Hz, 1H), 7.76 (d, J = 8.4 Hz, 1H), 7.42 (dd, J = 8.4Hz, 2.1 Hz, 1H), 7.36-7.31 (m, 2H), 7.00-6.96 (m, 1H), 5.22 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 152.00, 139.47, 136.86, 134.09, 132.21, 131.31, 131.06, 126.76, 125.59, 123.56, 123.51, 120.09, 117.91, 108.14. 高分辨质谱(EI模式):m/z for C14H7BrCl2OS [M]+ 计算值371.8778, 实测值 371.8772。
2)步骤1)所得产物 (8.4 mg, 0.022 mmol),碳酸钙(10 mg, 0.1 mmol)分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 溴化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4l (5 mg, 0.017 mmol, 77% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.98 (d, J = 1.5 Hz, 1H), 7.79 (d, J = 8.7 Hz, 1H), 7.72 (d, J = 2.1 Hz, 1H), 7.57 (d, J = 9.0 Hz, 1H), 7.39-7.35 (m, 2H). 13C NMR (101 MHz, 氘代氯仿): δ 157.22, 153.01, 140.17, 131.56, 129.19, 125.83, 125.75, 125.55, 125.35, 124.98, 119.67, 119.51, 113.64. 高分辨质谱(EI模式):m/z for C14H6Cl2OS [M]+ 计算值291.9516, 实测值 291.9518。
实施例13 3-氯-8-氟苯并[4,5]噻吩并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯5-氯苯并[b]噻吩(160 mg, 0.5 mmol)、5-氟-2-羟基苯硼酸(90 mg, 0.5 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (276 mg,2.0 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得2-(3-溴-5-氯苯并[b]噻吩-2-基)-4-氟苯酚3m, 产率为81%。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.87 (d, J = 1.5 Hz, 1H), 7.76 (d, J = 8.7 Hz, 1H), 7.42 (dd, J = 8.4, 1.8 Hz, 1H), 7.13-7.07 (m, 2H), 7.01-6.96 (m, 1H), 5.05 (s, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 158.13, 154.95, 149.47 (d, J = 2.2 Hz), 139.50, 136.82, 134.44, 132.18, 126.73, 123.52 (d, J = 3.4 Hz), 119.48 (d, J = 8.4 Hz), 118.32, 118.13-117.35 (m), 107.98. 高分辨质谱(EI模式):m/zfor C14H7BrClFOS [M]+ 计算值355.9074, 实测值 355.9076。
2)步骤1)所得产物 (26 mg, 0.072 mmol),碳酸氢钠(4 当量)分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),1,10-菲啰啉(0.1当量),90 oC反应15 min, 柱色谱分离得到目标化合物4m (13 mg, 0.047 mmol, 65% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.95 (d, J = 1.8 Hz, 1H), 7.77 (d, J = 8.7 Hz, 1H), 7.56 (dd, J = 9.0, 4.2 Hz, 1H), 7.40-7.34 (m, 2H), 7.15-7.08 (m, 1H). 13C NMR (75 MHz, 氘代氯仿): δ 160.92, 157.74, 155.01, 153.38, 140.10, 131.49, 125.78 (d, J = 18.8 Hz), 125.32, 124.49, 120.11, 119.63, 113.44-113.13 (m), 112.78, 105.76 (d, J = 25.5 Hz). 高分辨质谱(EI模式):m/z for C14H6ClFOS [M]+ 计算值275.9812, 实测值 275.9809。
实施例14 DTBDF的合成
1)准确称取2,3-二溴噻吩(2.0 mmol)、2,4-二羟基对苯二硼酸(1.0 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (552 mg,4.0 mmol),四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到中间产物3n,产率为 45%。1H NMR (400 MHz, 氘代二甲亚砜): δ 9.40 (brs, 1H), 7.70 (d, J = 5.4 Hz, 1H), 7.14 (d, J = 5.4 Hz, 1H), 6.94 (s, 1H). 13C NMR (101 MHz, 氘代二甲亚砜): δ 147.54, 134.69, 130.57, 127.57, 120.67, 118.72, 109.30. 高分辨质谱(EI模式):m/z for C14H8Br2O2S2 [M]+ 429.8332, 实测值 431.8315。
2)步骤1)所得产物 碳酸氢钠 (60 mg, 0.15 mmol),碳酸氢钠 (26 mg, 2 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(2 mL)中鼓泡除氧10 min, 碘化亚铜 (12 mg, 0.06 mmol),4,4'-二叔丁基-2,2'-联吡啶 (32 mg, 0.12 mmol),90 oC反应24 h, 柱色谱分离得到目标化合物4n (19 mg, 0.07 mmol, 47% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.80 (s, 2H), 7.41 (d, J = 5.3 Hz, 2H), 7.19 (d, J = 5.3 Hz, 2H). 高分辨质谱(EI模式):m/z for C14H6O2S2 [M]+ 计算值269.9809, 实测值 269.9807。
实施例15 DBFTT的合成
1)准确称取2,3,4,5-四溴噻吩(400 mg, 1.0 mmol)、2-羟基苯硼酸(414 mg, 3.0 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (552 mg,4.0 mmol)、四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到中间产物3o,产率为 49%。1H NMR (400 MHz, 氘代二甲亚砜): δ 10.08 (s, 2H), 7.41 (dd, J = 7.6, 1.5 Hz, 2H), 7.37-7.26 (m, 2H), 7.00 (d, J = 8.2 Hz, 2H), 6.93 (dd, J = 10.9, 4.1 Hz, 2H). 13C NMR (101 MHz, 氘代二甲亚砜): δ155.79, 138.19, 136.79, 132.18, 131.34, 119.51, 119.15, 116.61, 102.44. 高分辨质谱 (ESI) m/z for C18H10Br2O2S2[M]+ 计算值479.8489, 实测值 479.8483。
2)步骤1)所得产物 (0.5 mmol),碳酸钾 (2 mmol) 分别在氮气保护状态下加入到N,N-二甲基甲酰胺(5 mL)中鼓泡除氧10 min, 碘化亚铜(0.05当量),4,4'-二叔丁基-2,2'-联吡啶 (20 mg, 0.075 mmol, 10.05当量),90 oC反应15 min, 柱色谱分离得到目标化合物4o (42 mg, 0.13 mmol, 48% 产率)。该化合物的表征数据如下:1H NMR (300 MHz, 氘代氯仿): δ 7.71 (dd, J = 6.0, 3.0 Hz, 1H), 7.62 (dd, J = 6.2, 3.1 Hz, 1H), 7.41-7.33 (m, 2H). 高分辨质谱(EI模式):m/z for C18H8O2S2 [M]+ 319.9966, 实测值 319.9970。
实施例16 C8-DTBDF的合成
DTBDF (4n) (30 mg, 0.11 mmol) 溶解于 二氯甲烷 (2 ml) 中, 将上述溶液冷至-20 oC,然后加入氯化铝(65 mg, 0.5 mmol). 将反应混合液-78 oC, 然后逐滴加入辛酰氯 (97 mg, 102 , 0.6 mmol)。逐渐升至室温后,搅拌 24 h。反应完成后,于冰水浴中逐滴加入水淬灭反应。 用二氯甲烷萃取,无水硫酸镁干燥,过滤,减压下除去溶剂,得中间产物(不纯)。AlCl3悬浮于乙醚中,冷至-20 oC,然后加入 LiAlH4,搅拌 20 min。将上述中间产物溶于二氯甲烷 中,然后滴加到上述悬浊液中, 逐渐升至室温后,搅拌12 h. 反应完成后,于冰水浴中逐滴加入水淬灭反应。用正己烷萃取,无水硫酸镁干燥,过滤,减压下除去溶剂。硅胶柱层析分离,得白色固体状C8-DTBDF (35 mg, 0.07 mmol, 64%产率)。该化合物的表征数据如下:1H NMR (400 MHz, 氘代氯仿): δ 7.66 (s, 2H), 6.89 (s, 2H), 2.91 (t, J = 7.6 Hz, 4H), 1.80-1.69 (m, 4H), 1.44-1.22 (m, 10H), 0.88 (t, J = 6.8 Hz, 6H). 13C NMR (101 MHz, 氘代氯仿): δ 159.08, 155.24, 149.03, 120.86, 116.68, 109.02, 101.09, 31.86, 31.71, 31.63, 29.33, 29.22, 29.05, 22.66, 14.09. 高分辨质谱(EI模式):m/z for C30H38O2S2 [M]+ 计算值494.2313, 实测值 494.2308。
实施例17 苯并呋喃并[3,2-b]苯并呋喃的合成
1)准确称取2,3-二溴苯并[b]呋喃(208 mg, 1.0 mmol)、2-羟基苯硼酸(207 mg, 1.5 mmol),并依次加入到 25 mL 的舒伦克瓶中,加入碳酸钾 (552 mg, 4.0 mmol),四(三苯基膦)钯 (0.05当量),1,4-二氧六环/水 (体积比4:1),置于90 ℃油浴中反应6小时。反应结束后,减压除去溶剂,使用硅胶柱分离,石油醚/乙酸乙酯作为洗脱剂,得到2-(3-溴苯并[b]呋喃-2-基)苯酚 3p,产率为 70%。1H NMR (300 MHz, 氘代氯仿) δ 7.89 (dd, J = 7.8, 1.6 Hz, 1H), 7.55-7.45 (m, 1H), 7.45-7.37 (m, 1H), 7.37-7.22 (m, 3H), 7.04-6.96 (m, 2H), 6.94 (s, 1H). 13C NMR (75 MHz, 氘代氯仿) δ 154.26, 152.88, 149.39, 131.63, 129.58, 128.70, 125.99, 124.13, 120.50, 120.14, 117.61, 114.97, 111.33, 95.58.
2)步骤1)所得产物(70 mg, 0.333 mmol),噻吩-2-甲酸亚铜(70 mg, 0.366 mmol),分别在氮气保护状态下加入到N-甲基吡咯烷酮(1 mL)中, 150 oC反应1 h, 柱色谱分离,四氯化碳作为洗脱剂,得到目标化合物4p (10 mg, 0.048 mmol, 14% 产率)。该化合物的表征数据如下:1H NMR (400 MHz, 氘代氯仿): δ 7.77-7.79 (m, 2 H), 7.34-7.38 (m, 4 H), 7.61-7.64 (m, 2 H). 13C NMR (101 MHz, 氘代氯仿): δ 159.0, 144.2, 124.5, 123.5, 117.6, 117.4, 113.1。MS-APCI (+): mz 208 [M]+.
实施例18 利用电化学循环伏安法测量C8-DTBDF最高占据分子轨道
利用电化学工作站对实施例16所得产物C8-DTBDF的电化学特性进行了测试,以二茂铁为内标,电解液为六氟磷酸四丁基胺的乙腈溶液(浓度为0.1M)。采用标准的三电极系统进行测试,以铂丝作为对电极、氯化银电极作为参比电极。测得其循环伏安曲线如图2所示,可算出C8-DTBDF的最高占据分子轨道能级为-5.42 eV。
实施例19 有机场效应器件性质分析
分别由高度掺杂的硅晶片与300 nm 二氧化硅作为栅极电极和介电层。通过剥离技术,30 nm的金电极沉积在基板上,形成长度范围从5−50 和宽度为1.4 mm的沟道。十八烷基三氯硅烷基修饰:在真空炉中,在120 ℃条件下加热3 h,处理的衬底依次用正己烷、乙醇和氯仿冲洗。
通过溶液甩膜的方法将半导体材料C8-DTBDF(5 mg/ml, 二氯甲烷溶液)旋涂到十八烷基三氯硅烷基修饰的二氧化硅 (300 nm)/硅基板上,形成底栅底接触的薄膜有机场效应器件,分别在80 °C和120 °C条件下进行退火处理。测定器件的转移曲线(见图3)。器件表现出p型特征,在80 °C退火后的器件特性最好,其迁移率最高可达6.9 × 10-4 cm2V-1s-1,阀值电压16 V和开关比可达106。以上结果表明DTBDF是具有发展潜力的骨架。
以上所述仅为本发明的较佳实施例而已,并不用以限制本发明,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!