含酰腙键的芳香族端羟基扩链剂、自修复聚氨酯及其制备方法与流程
本发明属于聚合物材料领域,具体涉及自修复聚氨酯,尤其涉及含酰腙键的芳香族端羟基扩链剂、自修复聚氨酯及其制备方法。
背景技术:
聚合物材料因其优异的性能而被广泛应用于航空、航天、电子、机械等高新技术领域。然而,材料在使用过程中不可避免会产生损伤,其中,微裂纹是材料微观损伤的主要形式,微裂纹的产生会导致材料力学性能下降从而降低材料使用寿命。自修复功能是模仿生物体损伤自修复的机理对材料内部微裂纹实现自修复,避免材料进一步破坏,延长材料使用寿命,为预防潜在的危害提供了一种新方法。
聚合物材料的自修复方法目前大致分为两大类,外援型自修复和本征型自修复。外援型自修复,即借助外加修复剂实现材料的自修复功能,即将包有修复剂的微胶囊、液芯纤维或者毛细管网络等包覆材料埋植于基体中,外部损伤可诱发包覆材料破裂,释放出修复剂对裂缝修复。外援型自修复的修复方法简单、修复效率也较高。但外援型自修复存在以下问题:(1)只能实现对微裂缝的有限次自修复,至材料内部的修复剂释放完毕则失去自修复功能;(2)修复剂释放后产生的微小空隙将成为材料新的缺陷,不利于材料长期使用;(3)自修复材料中的微胶囊、液芯纤维和毛细管网络等修复单元不易均匀分散,易发生团聚,一定程度上影响修复效果。
本征型化学自修复,即将可逆化学键(可逆共价键和可逆非共价键)通过化学反应引入到聚合物本体中,通过加热、调节ph等方式促使体系中某些化学键的可逆反应,进而使链段相互连接来实现自修复。与外援型修复体系相比,本征型自修复体系具有以下优势:(1)不需要考虑外加物质与基体的相容性,自修复材料可以在物体受损时实现自我修复;(2)本征型自修复可实现对裂纹甚至断裂材料的多次自修复。此外,由于可逆共价键比可逆非共价键的键能高,因此,利用可逆共价键可以实现本征型自修复材料更好的稳定性,进而可得到力学性能较好的自修复材料。
近几年,基于可逆共价键的自修复聚氨酯材料取得了不错的进展,但各类自修复聚氨酯体系大多力学性能均有待改进。如基于可逆亚胺键的自修复聚氨酯水凝胶体系,多为软质材料且不具备拉伸条件;而基于diels-alder反应的自修复聚氨酯体系表现出硬、脆的特性。基于目前自修复聚氨酯体系的力学性能,极大地限制了自修复聚氨酯体系的应用。发明专利申请cn106117486a公开了“含酰腙键的二元醇、含酰腙键和双硫键的二元醇、自修复聚氨酯弹性体及其制备方法”。该申请制备的自修复聚氨酯弹性体在受到机械损伤后,聚合物交联分子链上的酰腙键或双硫键通过动态交换反应实现材料的自修复。其优势在于:(1)自修复交联聚氨酯弹性体材料的制备简单,原料易得;(2)在室温下即可实现自修复,修复能力高、修复时间短、可实现多次自修复,且具有较好的力学性能。然而,该申请中所述的自修复聚氨酯体系的软段为两种聚醚多元醇(ppg2000和tep-240),自修复聚氨酯体系的性能有待进一步提升,具体表现为力学性能较差、无支撑能力、拉伸强度为1.83mpa、断裂伸长率450.72%以及邵a硬度值为15,材料较软,因此大大限制了其应用。
聚酯多元醇的反应活性高、稳定性好,便于生产和操作。以聚酯多元醇为基础的聚氨酯材料,通常都具有力学机械性能好,耐油、抗磨性能优越等特点,使得聚酯多元醇应用领域较广,是制造胶黏剂、弹性体、涂料、油墨、密封胶、革用树脂、鞋用树脂、tpu等产品的重要材料。然而,将聚酯多元醇用于自修复聚氨脂体系,目前尚未见报道。
技术实现要素:
基于目前自修复聚氨酯材料的现状,本发明提供了一种含酰腙键的芳香族端羟基扩链剂、采用所述芳香族端羟基扩链剂制备的自修复聚氨酯及其制备方法。本发明所制备的自修复聚氨酯材料中,所述聚氨酯分子链含有可逆酰腙键,在室温下即可实现自修复,不但修复效率高,同时具有较好的力学性能。此外,所述自修复聚氨酯制备方法简单,原料均为已商业化生产的产品,聚氨酯材料性能可控性强,具有广泛的应用前景。
本发明的技术方案:
含酰腙键的芳香族端羟基扩链剂,结构通式为式(i)或式(ii)所示:
其中,r1选自烷基、芳香基或取代的芳香基;r2选自氢、烷基、烷氧基、氨基、羟基、芳香基或取代的芳香基;r3选自烷基、环烷基、芳香基或取代的芳香基。例如:对苯二甲酸二甲酰肼+4-羟基-4-甲基-2-戊酮。
其中,r4选自芳香基或取代的芳香基;r5选自氢、烷基、烷氧基、羟基;r6选自芳香基或取代的芳香基,n1+n2+n3≥2,保证扩链剂的分子结构至少含有两个端羟基,即包括以下几种情形:(1)两个端羟基,都在左侧;(2)两个端羟基,都在右侧;(3)两个端羟基,左右各一个;(4)两个以上羟基,左右两侧都有。
与含酰腙键的二元醇相比,本发明所述的含酰腙键的芳香族端羟基扩链剂的分子结构中存在酰肼苯环的大π键和碳氧双键的π键的π-π共轭效应,使该扩链剂结构稳定。
含酰腙键的芳香族端羟基扩链剂,按重量份数计,由以下原料反应得到:16-42份的酰肼化合物和12-35份的α-羟基羰基化合物。其制备方法包括以下步骤:(1)按重量份数计,将12-35份的α-羟基羰基化合物溶于去离子水或有机溶剂中;将16-42份的酰肼化合物溶于冰乙酸中,混合;所述有机溶剂为二甲基亚砜、n,n-二甲基甲酰胺、异丙醇、丙酮或乙酸乙酯;所述去离子水/有机溶剂和冰乙酸的重量份数之和为120份。(2)在50℃-70℃的温度条件下反应2-4h,得到含酰腙键的芳香族端羟基扩链剂。
其中,所述的酰肼类化合物为苯酰肼、苄氧基甲酰肼、3-苄氧基苯甲酰肼、4-苄氧基苯甲酰肼、3-氨基苯酰肼、3-羟基苯酰肼、水杨酰肼、4-羟基苯甲酰肼、3-乙氧基苯酰肼、4-苯氧基苯酰肼、4-氯苯甲酰肼、2,4-二氯苯酰肼、2,5-二氯苯酰肼、2,5-二甲氧基苯酰肼、2,4,6-三氯苯肼、3,5-二羟基苯酰肼、甲基二甲酰肼、癸二酸二酰肼、对苯二甲酸二酰肼、邻苯二甲酸二酰肼、乙二酸二酰肼、马来酸二酰肼、丁二酸二酰肼、己二酸二酰肼和壬二酸二酰肼中的任一种或多种的混合物。
其中,所述的α-羟基羰基化合物为对羟基苯甲醛、水杨醛、水杨酸、2-羟基肉桂醛、2,5-二羟基苯甲醛、3-羟基苯乙酮、4-羟基-2-丁酮、4-羟基-3-己酮、4-羟基环己酮、4-羟基-4-甲基-2-戊酮、4-羟基-3-甲基苯乙酮、2-羟基-5-甲基苯乙酮、4-氨基-3羟基苯甲酸和4-氨基-3硝基苯甲酸中的任一种或多种的混合物。
利用所述的芳香族端羟基扩链剂制备的自修复交联聚氨酯,通过以下方法制备:(1)按重量份数计,将2-7份所述的含酰腙键的芳香族端羟基扩链剂溶于6-15份的二甲基亚砜中;(2)加入20-50份的端异氰酸酯基的聚氨酯预聚体,在50℃-80℃下搅拌反应4h-12h后抽真空脱除气泡,25℃-60℃下固化6-24h,得到自修复交联聚氨酯;(3)将聚氨酯浸没在去离子水中,浸没的温度条件为50℃-80℃,浸没的时间为12h-24h;(4)取出浸没后的聚氨酯并干燥,直至聚氨酯重量恒定;(5)重复步骤(3)和(4)两次,得到不含溶剂的自修复交联聚氨酯。
其中,所述端异氰酸酯基的聚氨酯预聚体通过以下方法制备:按重量份数计,在50℃-80℃的温度条件下,向50-120份的聚醚-聚酯多元醇混合物与11-34份的二异氰酸酯中,加入适量催化剂二月桂酸二丁基锡,反应1h-6h,得到端异氰酸酯基的聚氨酯预聚体。所述二月桂酸二丁基锡的用量为0.35-0.80重量份;所述聚醚-聚酯多元醇混合物中聚醚多元醇和聚酯多元醇的摩尔为1:2-1:7;所述聚醚和聚酯多元醇的数均分子量均为400-10000。所述的二异氰酸酯为甲苯-2,4-二异氰酸酯(tdi)、二苯基甲烷-2,2′-二异氰酸酯(mdi)、二苯基甲烷-2,4′-二异氰酸酯、二苯基甲烷-4,4′-二异氰酸酯、六亚甲基二异氰酸酯(hdi)和异佛尔酮二异氰酸酯(ipdi)中的任一种或多种的混合物。本发明采用中的自修复聚氨酯弹性体体系综合了聚醚多元醇和聚酯多元醇的特点,经过相应的软硬段调节,使材料的拉伸强度和断裂伸长率得到同时提高,其中,拉伸强度提高至2.75mpa,邵氏a硬度值提高至20,使本发明中的自修复聚氨酯材料具有较广阔的应用前景。这是由于,聚酯多元醇制成的聚氨酯含极性大的酯基,使聚氨酯体系内部不仅硬段间能够形成氢键,而且软段上的极性基团也能部分地与硬段上的极性基团形成氢键,使硬相能更均匀地分布于软相中,起到弹性交联点的作用。
本发明的有益效果:与现有自修复聚氨酯相比,本发明的有益效果:
(1)本发明利用含酰腙键的芳香族端羟基扩链剂与端异氰酸酯基的端异氰酸酯基聚氨酯预聚体反应,合成出分子链含可逆酰腙键的自修复聚氨酯,合成的聚氨酯除去溶剂后得到本体聚合物,故具有更好的物理机械性能,且可实现室温下自修复且切开的横断面在室温或酸性条件下,接触24h即可实现自修复,自修复效率高。
(2)相比于普通含酰腙键的端羟基扩链剂,所述含酰腙键的芳香族端羟基扩链剂具有稳定的共轭结构,来自于分子结构中酰肼苯环的大π键和碳氧双键的π键所形成的π-π共轭效应,使该扩链剂结构稳定;同时,所述含酰腙键的芳香族端羟基扩链剂与所述自修复聚氨酯体系的相容较好,可形成更加紧密的聚氨酯交联结构。
(3)所述的聚氨酯体系的软段特性由聚酯多元醇和聚醚多元醇共同提供,与其他单一的聚酯多元醇或聚醚多元醇聚氨酯体系相比,材料拉伸强度和断裂伸长率均有提高;通过调整合适的配方比,两种多元醇与相应异氰酸酯反应得到端异氰酸酯基的聚氨酯预聚物,预聚物的粘度适中,利用后续合成反应的进行;最终得到具有较好力学性能的聚氨酯弹性体材料,该聚氨酯体系所需的原料易于获取,制备工艺方法简单,材料性能可控性强。
说明书附图
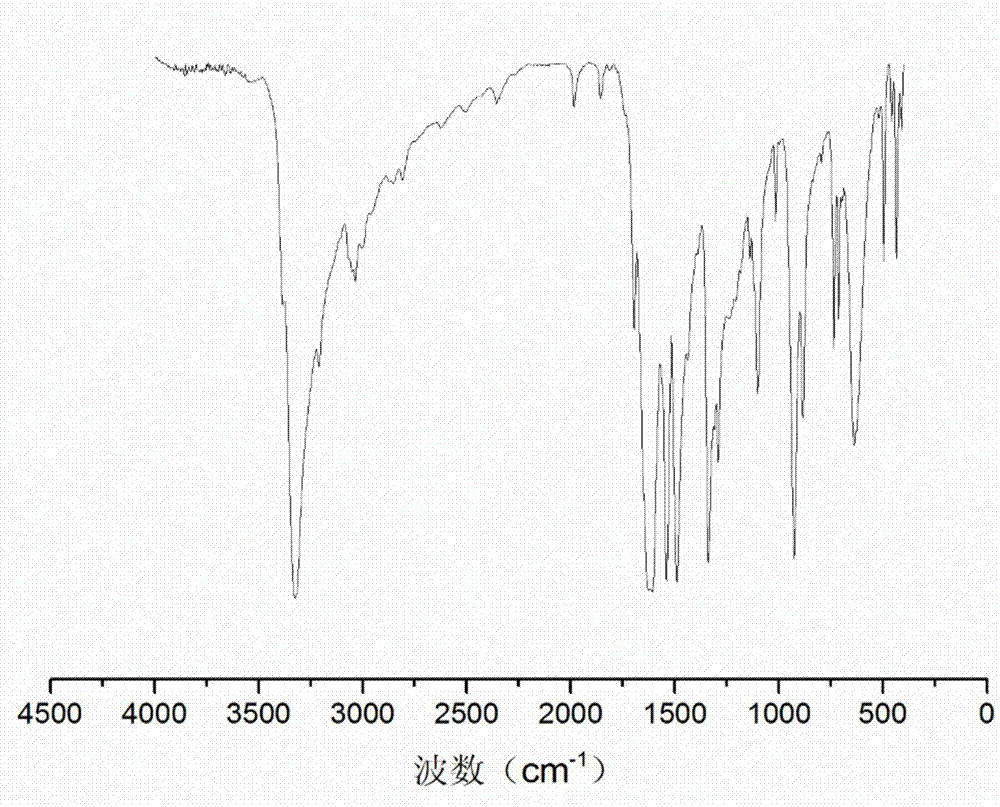
图1为本发明实施例1中酰肼化合物对苯二甲酸二甲酰肼的红外谱图;
图2为本发明实施例1中酰肼化合物对苯二甲酸二甲酰肼的核磁氢谱谱图;
图3为本发明实施例1中含酰腙键的芳香族端羟基扩链剂4-羟基-4-甲基-2-戊酮缩对苯二甲酸二甲酰腙的红外谱图;
图4为本发明实施例1中含酰腙键的芳香族端羟基扩链剂4-羟基-4-甲基-2-戊酮缩对苯二甲酸二甲酰腙的核磁氢谱谱图。
具体实施方式
下面结合实施例对本发明做进一步的说明。
实施例1.1:制备含酰腙键的芳香族端羟基扩链剂
取23.23g4-羟基-4-甲基-2-戊酮溶解在50.00g去离子水中,取19.42g对苯二甲酸二甲酰肼溶解在70.00g冰乙酸中。将两者混合均匀后在70℃,250rpm搅拌下反应2h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(1)所示,命名为4-羟基-4-甲基-2-戊酮缩对苯二甲酸二甲酰腙,产物为白色固体粉末。获得产物33.20g,产率85%,其红外光谱图如图3所示,其核磁氢谱谱图如图4所示。
实施例1.2:制备含酰腙键的芳香族端羟基扩链剂
取13.81g2,5-二羟基苯甲醛溶解在90.00g异丙醇中,取16.82g3,5-二羟基苯酰肼溶解在30.00g冰乙酸中。将两者混合均匀后在50℃,250rpm搅拌下反应4h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为2,5-二羟基苯甲醛缩3,4-二羟基苯酰腙,产物为淡黄色固体粉末。获得产物23.72g,产率82.3%。
实施例1.3:制备含酰腙键的芳香族端羟基扩链剂
取27.62g水杨酸溶解在70.00g去离子水中,取42.00g3,5-二羟基苯酰肼溶解在50.00g冰乙酸中。将两者混合均匀后在65℃,250rpm搅拌下反应2.5h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为水杨酸缩3,5-二羟基苯酰腙,产物为淡黄色固体粉末。获得产物44.68g,产率77.5%。
实施例1.4:制备含酰腙键的芳香族端羟基扩链剂
取22.22g2-羟基肉桂醛溶解在60.00g异丙醇中,取22.82g水杨酰肼溶解在60.00g冰乙酸中。将两者混合均匀后在60℃,250rpm搅拌下反应3h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为2-羟基肉桂醛缩水杨酰腙,产物为黄色固体粉末。获得产物30.01g,产率70.87%。
实施例1.5:制备含酰腙键的芳香族端羟基扩链剂
取24.42g对羟基苯甲醛溶解在30.00g异丙醇中,取26.42g马来酸二酰肼溶解在90.00g冰乙酸中。将两者混合均匀后在55℃,250rpm搅拌下反应3.5h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为对羟基苯甲醛缩马来酸二酰腙,产物为淡黄色固体粉末。获得产物37.52g,产率79.43%。
实施例1.6:制备含酰腙键的芳香族端羟基扩链剂
取35.00g2,5-二羟基苯甲醛溶解在60.00g去离子水中,取41.55g苄氧基甲酰肼溶解在60.00g冰乙酸中。将两者混合均匀后在50℃,250rpm搅拌下反应2h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为2,5-二羟基苯甲醛缩苄氧基甲酰腙,产物为黄色固体粉末。获得产物59.9g,产率83.7%。
实施例1.7:制备含酰腙键的芳香族端羟基扩链剂
取12.00g水杨酸溶解在40.00g去离子水中,取16.00g苄氧基甲酰肼溶解在80.00g冰乙酸中。将两者混合均匀后在68℃,250rpm搅拌下反应2h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为水杨酸缩苄氧基甲酰腙,产物为淡黄色固体粉末。获得产物22.91g,产率80.02%。
实施例1.8:制备含酰腙键的芳香族端羟基扩链剂
取24.42g对羟基苯甲醛溶解在50.00g异丙醇中,取38.04g4-羟基苯甲酰肼溶解在70.00g冰乙酸中。将两者混合均匀后在58℃,250rpm搅拌下反应2h。减压过滤,将所得产物真空干燥,即得到含酰腙键的芳香族端羟基扩链剂,分子结构式如式(2)所示,命名为对羟基苯甲醛缩4-羟基苯甲酰腙,产物为黄色固体粉末。获得产物43.21g,产率84.3%。
实施例2.1:制备端异氰酸酯基的聚氨酯预聚体
取56.50g弹性体聚醚tep-240与聚己内酯二元醇(pcl210)的混合物、13.76g2,4-二苯基甲烷二异氰酸酯与4,4’-二苯基甲烷二异氰酸酯的混合物(mdi-50)、0.35gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,弹性体聚醚tep-240与聚己内酯二元醇(pcl210)的摩尔比为1:4。
上述反应物在65℃,250rpm搅拌下抽真空反应4h,得到异氰酸酯基封端预聚物mdi50-tep240-pcl210-mdi50,此时预聚物体系中异氰酸酯基含量为3.27wt%。
实施例2.2:制备端异氰酸酯基的聚氨酯预聚体
取80.00g聚乙二醇peg2000与聚己内酯二元醇(pcl220)的混合物、13.93g甲苯-2,4-二异氰酸酯tdi、0.47gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,聚乙二醇peg2000与聚己内酯二元醇(pcl220)的摩尔比为1:3。
上述反应物在65℃,250rpm搅拌下抽真空反应4h,得到异氰酸酯基封端预聚物tdi-peg2000-pcl220-tdi,此时预聚物体系中异氰酸酯基含量为3.56wt%。
实施例2.3:制备端异氰酸酯基的聚氨酯预聚体
取66.50g弹性体聚醚tep-240与聚丙交酯二元醇(pla210m)的混合物、18.77g二苯基甲烷-2,2′-二异氰酸mdi、0.43gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,弹性体聚醚tep-240与聚丙交酯二元醇(pla210m)的摩尔比为1:6。
上述反应物在75℃,250rpm搅拌下抽真空反应2h,得到异氰酸酯基封端预聚物mdi-tep240-pla210m-mdi,此时预聚物体系中异氰酸酯基含量为3.68wt%。
实施例2.4:制备端异氰酸酯基的聚氨酯预聚体
取70.75g弹性体聚醚tep-240与聚碳酸酯二元醇(pc230)的混合物、15g二苯基甲烷-2,4′-二异氰酸酯mdi、0.43gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,弹性体聚醚tep-240与聚碳酸酯二元醇(pc230)的摩尔比为1:7。
上述反应物在80℃,250rpm搅拌下抽真空反应1h,得到异氰酸酯基封端预聚物mdi-tep240-pc230-mdi,此时预聚物体系中异氰酸酯基含量为2.92wt%。
实施例2.5:制备端异氰酸酯基的聚氨酯预聚体
取80.00g聚丙二醇ppg2000与聚己内酯二元醇(pcl210)的混合物、30.03g二苯基甲烷-4,4′-二异氰酸酯(mdi)、0.55gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,聚丙二醇ppg2000与聚己内酯二元醇(pcl210)的摩尔比为1:2。
上述反应物在50℃,250rpm搅拌下抽真空反应6h,得到异氰酸酯基封端预聚物mdi-ppg2000-pcl210-mdi,此时预聚物体系中异氰酸酯基含量为4.56wt%。
实施例2.6:制备端异氰酸酯基的聚氨酯预聚体
取120.00g聚丙二醇ppg2000与聚丙交酯二元醇(pla210m)的混合物、34.00g六亚甲基二异氰酸酯(hdi)、0.80gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,聚丙二醇ppg2000与聚丙交酯二元醇(pla210m)的摩尔比为1:4。
上述反应物在55℃,250rpm搅拌下抽真空反应5h,得到异氰酸酯基封端预聚物hdi-ppg2000-pla210m-hdi,此时预聚物体系中异氰酸酯基含量为5.44wt%。
实施例2.7:制备端异氰酸酯基的聚氨酯预聚体
取65.00g聚四氢呋喃醚二醇(ptmeg1000)与聚碳酸酯二元醇(pc230)的混合物、11.00g异佛尔酮二异氰酸酯(ipdi)、0.38gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,聚四氢呋喃醚二醇(ptmeg1000)与聚碳酸酯二元醇(pc230)的摩尔比为1:4。
上述反应物在60℃,250rpm搅拌下抽真空反应4h,得到异氰酸酯基封端预聚物ipdi-ptmeg1000-pc230-ipdi,此时预聚物体系中异氰酸酯基含量为2.74wt%。
实施例2.8:制备端异氰酸酯基的聚氨酯预聚体
取50.00g聚四氢呋喃醚二醇(ptmeg1000)与聚己内酯二元醇(pcl210)的混合物、17.41g甲苯-2,4-二异氰酸酯tdi、0.35gdbtdl(二月桂酸二丁基锡)于三口烧瓶中;其中,聚四氢呋喃醚二醇(ptmeg1000)与聚己内酯二元醇(pcl210)的摩尔比为1:4。
上述反应物在65℃,250rpm搅拌下抽真空反应4h,得到异氰酸酯基封端预聚物tdi-ptmeg1000-pcl210-tdi,此时预聚物体系中异氰酸酯基含量为6.19wt%。
实施例3.1:制备自修复聚氨酯
取4.56g实施例1.1制备的含酰腙键的芳香族端羟基扩链剂4-羟基-4-甲基-2-戊酮缩对苯二甲酸二甲酰腙溶解于8.00g二甲基亚砜中,加入30.00g实施例2.1制备的聚氨酯预聚体,在60℃下搅拌反应(搅拌桨转速250rpm)10h后,抽真空30min脱除气泡;浇注于敞口模具中,在60℃下固化成型6h。浸没在50倍质量的去离子水中,在50℃下浸没24h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.2:制备自修复聚氨酯
取2.00g实施例1.2制备的含酰腙键的芳香族端羟基扩链剂2,5-二羟基苯甲醛缩3,4-二羟基苯酰腙溶解于7.00g二甲基亚砜中,加入35.00g实施例2.2制备的聚氨酯预聚体,65℃下搅拌反应(搅拌桨转速250rpm)12h后抽真空30min脱除气泡;,浇注于敞口模具中,在25℃下固化成型9h。浸没在50倍质量的去离子水中,在55℃下浸没22h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.3:制备自修复聚氨酯
取2.53g实施例1.3制备的含酰腙键的芳香族端羟基扩链剂水杨酸缩3,5-二羟基苯酰腙溶解于6.00g二甲基亚砜中,加入40.00g实施例2.3制备的聚氨酯预聚体,70℃下搅拌反应(搅拌桨转速250rpm)4h后抽真空30min脱除气泡;,浇注于敞口模具中,在30℃下固化成型12h。浸没在50倍质量的去离子水中,在60℃下浸没20h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.4:制备自修复聚氨酯
取4.42g实施例1.4制备的含酰腙键的芳香族端羟基扩链剂2-羟基肉桂醛缩水杨酰腙溶解于12.00g二甲基亚砜中,加入45.00g实施例2.4制备的聚氨酯预聚体,75℃下搅拌(搅拌桨转速250rpm)反应6h后抽真空30min脱除气泡;浇注于敞口模具中,在35℃下固化成型15h。浸没在50倍质量的去离子水中,在65℃下浸没18h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.5:制备自修复聚氨酯
取6.41g实施例1.5制备的含酰腙键的芳香族端羟基扩链剂对羟基苯甲醛缩马来酸二酰腙溶解于15.00g二甲基亚砜中,加入50.00g实施例2.5制备的聚氨酯预聚体,80℃下搅拌(搅拌桨转速250rpm)反应8h后抽真空30min脱除气泡;,浇注于敞口模具中,在40℃下固化成型18h。浸没在50倍质量的去离子水中,在70℃下浸没16h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.6:制备自修复聚氨酯
取3.71g实施例1.6制备的含酰腙键的芳香族端羟基扩链剂2,5-二羟基苯甲醛缩苄氧基甲酰腙溶解于9.00g二甲基亚砜中,加入20.00g实施例2.6制备的聚氨酯预聚体,50℃下搅拌(搅拌桨转速250rpm)反应5h后抽真空30min脱除气泡;浇注于敞口模具中,在45℃下固化成型21h。浸没在50倍质量的去离子水中,在75℃下浸没14h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.7:制备自修复聚氨酯
取2.33g实施例1.7制备的含酰腙键的芳香族端羟基扩链剂水杨酸缩苄氧基甲酰腙溶解于10.00g二甲基亚砜中,加入25.00g实施例2.7制备的聚氨酯预聚体,55℃下搅拌(搅拌桨转速250rpm)反应7h后抽真空30min脱除气泡;,浇注于敞口模具中,在50℃下固化成型24h。浸没在50倍质量的去离子水中,在80℃下浸没12h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例3.8:制备自修复聚氨酯
取7.00g实施例1.8制备的含酰腙键的芳香族端羟基扩链剂对羟基苯甲醛缩4-羟基苯甲酰腙溶解于14.00g二甲基亚砜中,加入35.00g实施例2.8制备的聚氨酯预聚体,60℃下搅拌(搅拌桨转速250rpm)反应9h后抽真空30min脱除气泡;浇注于敞口模具中,在55℃下固化成型15h。浸没在50倍质量的去离子水中,在70℃下浸没16h,将二甲基亚砜置换出来;在110℃下真空干燥,直至聚氨酯重量不再变化,如此重复浸泡与烘干步骤两次后得到本发明所述的不含溶剂的交联的自修复聚氨酯。
实施例4:采用拉伸实验表征材料力学性能及自修复效率
将实施例3.1-3.8制备的聚氨酯按照gb/t528-2009制备成哑铃型拉伸样条。在样条中部沿垂直于拉伸轴线的方向将样条切断。室温下,含可逆酰腙键的聚氨酯样条沿切开的横断面接触在一起并在断面接触面的缝隙处滴加2滴冰乙酸,作为催化可逆价键自修复的催化剂,然后在样条上方施加5n的力,从而使切断的样条紧密接触在一起,接触24h后即可实现自修复。拉伸速率为100mm/min。自修复效率可用以下两个物理量的比值表示:
其中,σ0、σ1分别为自修复前与自修复后的拉伸强度;ε0、ε1分别为自修复前和自修复后的断裂伸长率。
表1.实施例3.1-3.8制备的自修复聚氨酯表征结果
根据上述表征结果可知,与现有技术相比,本发明所制备的自修复聚氨酯的拉伸强度最大提高至2.75mpa(实施例3.1),最小提高至2.64mpa(实施例3.8),断裂伸长率最大提高至495.37%(实施例3.7),最小提高至468.76%(实施例3.3),硬度最大提高至邵a20(实施例3.1),最小提高至邵a17(实施例3.8)。上述结果充分说明,本发明制备的自修复聚氨酯不但原料易于获取、制备工艺方法简单、材料性能可控性强,而且具备良好的力学性能,具有广阔的市场应用前景。
- 还没有人留言评论。精彩留言会获得点赞!