由缔合型共聚物表面改性而成的纤维素微丝及包含其的增稠剂组合物的制作方法
本发明涉及由通过共聚亲水性单体及疏水性单体获得的缔合型共聚物表面改性而成的纤维素微丝、包含其的增稠剂组合物及其的制备方法。
背景技术:
通常,增稠剂作为添加剂包含在如化妆品组合物等的体系中,用于调节操作性及流变性等。合成聚合物增稠剂用于提高体系的粘度,并防止水相和油相的相分离,从而用于增强复合流体的相稳定度。用作增稠剂的有机化合物有丙烯酸聚合物、羧基乙烯基聚合物等的合成聚合物,作为增稠剂的无机化合物有包含蒙脱石在内的各种粘土矿物和二氧化硅等。近来,对于包括多个合成表面活性剂的效果的天然化妆品的关注全球范围内趋于增加,以欧洲为中心,对有机/天然化妆品进行认证的机关开展活动。尤其,源自石油的聚乙二醇(peg)附加合成表面活性剂和合成聚合物的增稠剂的使用趋于严格,因此正在活跃进行着对能够代替其的天然聚合物及天然增稠剂的研究。
合成聚合物增稠剂因规定的分子量和聚合物链的扭曲现象而呈现均匀的增稠效果。温度变化、盐的投入、ph变化等的多种外部条件下也保持比较稳定的水溶液相。具有代表性的卡波姆(carbomer)最广为利用,并且包括卡波姆在内的大部分的合成聚合物增稠剂可通过溶解或分散于水中来用作增稠剂。通常,相对于化妆品组合物的总重量,添加0.001~20wt%的增稠剂,并且可直接调配于化妆品组合物中或用水或溶剂稀释成适当的粘度之后来进行调配。然而,由于对低刺激、无刺激产品的需求稳步增加,因此不仅在通过增稠剂确保剂型稳定性的方面上,而且还在使用感的差别化和赋予皮肤安全感的观点上,逐渐趋向于要求替代为环保型及天然原料,在相关领域中对于这种天然增稠剂及表面活性剂的技术开发持续进行着。
近年来,随着包括化妆品工业在内的整个衣食住方面上对于环保型、天然原料的关注进一步提高,对发挥与现有的增稠剂相似的性能的天然增稠剂的要求也提高,然而在替代基于合成聚合物的增稠剂的方面上还存在困难。进而,现有的合成聚合物增稠剂在制备工序中使用大量的有机溶剂、化学单体、化学引发剂、聚合抑制剂等,并在未反应单体的去除和中和过程中也再次利用大量的有机溶剂。这种有机物不仅被归类为对环境有害的物质,还包括引起皮肤毒性的可能性。并且,现有的合成聚合物掺入化妆品组合物中时,在配制成规定浓度以上的情况下,存在涂敷于皮肤时获得干涩和黏乎等让人不满意的使用感的问题。作为天然物质,以黄原胶、硬葡聚糖等作为原料的研究正在进行,但存在无法表现出与现有的增稠剂相似的增稠效果或使用感不好的缺点。
韩国公开专利公报第2001-0075666号涉及缔合型增稠剂的混合物及水性保护涂敷组合物,作为缔合型增稠剂,公开有改性为疏水性的纤维素衍生物。然而,所公开的羟乙基纤维素、甲基纤维素、羧甲基纤维素、乙基羟乙基纤维素等的纤维素衍生物因在纤维素表面仅具有疏水性残基,因此未呈现出较大的增稠效果。
对此本发明人为了开发出与现有合成增稠剂不同的源自天然的增稠剂,将缔合型共聚物适用聚合物接枝技术来引入纤维素的表面之后,进行氧化反应来制备了缔合型纤维素微丝的分散液,从而确认了其的作为增稠剂的非常优秀的效果并完成了本发明。
[现有技术文献]
专利文献
专利文献1:韩国公开专利公报第2001-0075666号
技术实现要素:
本发明要解决的技术问题
本发明的目的在于,提供由通过共聚亲水性单体及疏水性单体获得的缔合型共聚物表面改性而成的纤维素微丝。
本发明的再一目的在于,提供包含上述纤维素微丝的增稠剂组合物。
本发明的还一目的在于,提供包含上述增稠剂组合物的化妆品组合物。
本发明的另一目的在于,提供由缔合型共聚物表面改性而成的纤维素微丝的制备方法。
技术方案
本发明提供由通过共聚亲水性单体及疏水性单体获得的缔合型共聚物表面改性而成的纤维素微丝。
并且,本发明提供由缔合型共聚物表面改性而成的纤维素微丝的制备方法,其中,包括:步骤1),通过使包含纤维素、亲水性单体及疏水性单体的溶液进行活性自由基共聚反应来将缔合型共聚物接枝于纤维素表面;以及步骤2),通过添加2,2,6,6-四甲基哌啶-1-氧基(2,2,6,6-tetramethylpiperidine-1-oxyl)自由基引发剂来进行氧化反应并获得纤维素微丝。
有益效果
根据本发明的一具体例的纤维素微丝因由缔合型共聚物表面改性而成而纤维素微丝形成凝胶相。这种三维微丝凝胶相不仅因强大的水合行为而发挥优秀的增稠效果,还通过微丝之间的物理性相互作用而表现出区别化的流变行为。本发明的纤维素微丝结构上稳定,表现出耐化学性、耐盐性、耐酸碱性,并赋予区别化的使用感和皮肤安全性、化妆品剂型稳定性。因此,利用上述纤维素微丝的增稠剂组合物可广泛适用于化妆品组合物、皮肤外用药组合物等化妆品市场,并且预期在基于复合流体的多种工业中的利用度相当高。
附图说明
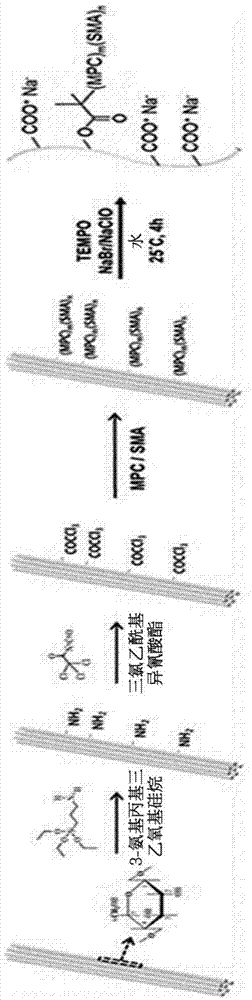
图1为示出根据本发明一实施例的由缔合型共聚物表面改性而成的纤维素微丝的制备过程的图。
图2为通过透射电子显微镜(tem)的纤维素微丝的形态学确认结果:(左)未进行表面改性的纤维素微丝,(右)引入poly(mpc-co-sma)的纤维素微丝。
图3为利用氢核磁共振(1h-nmr)的2-甲基丙烯酰氧乙基磷酸胆碱(mpc,2-methacryloyloxyethylphosphorylcholine)和甲基丙烯酸十八酯(sma,stearylmethacrylate)的合成效率分析结果,并确认了表面聚合时存在于溶剂中的单体的转化率:(左)合成前,(右)合成后的氢核磁共振(1h-nmr)数据。
图4为为了确认纤维素表面中合成了2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)而利用傅里叶变换红外光谱学(ft-ir)的定性分析结果。
图5为由缔合型共聚物表面改性而成的纤维素微丝分散液的增稠效果测定结果。
图6a至图6d为测定由缔合型共聚物表面改性而成的纤维素微丝分散液的流变特性的结果:按照各个甲基丙烯酸十八酯(sma)含量时,(a)弹性模量、(b)粘性模量、(c)剪应力、(d)屈服应力。
图7为由缔合型共聚物表面改性而成的纤维素微丝分散液的耐盐性及耐酸碱性的测定结果:(a)向未进行表面改性的微丝分散液(neatcnfs)和表面改性的微丝分散液(acnfs)添加0、1或1.5m的nacl之后,测定粘度并确认耐盐性的结果,(b)将表面改性的微丝分散液(neatcnfs)和表面改性的微丝分散液(acnfs)调节为ph2、7或11之后,测定粘度并确认耐酸碱性的结果。
具体实施方式
本发明一具体例提供由通过共聚亲水性单体及疏水性单体获得的缔合型共聚物表面改性而成的纤维素微丝。
在本说明书中所使用的术语,“缔合性(associative)”是指具有相同种类的分子借助两种以上的分子间力结合并形成(缔合)具有松弛的规则性的集合体的性质。
在本说明书中所使用的术语,“共聚(copolymerization)”是指通过混合、聚合两种或更多种的单体来合成共聚物(copolymer)的反应。当聚合a、b两种单体时,通常a和b不规则地混合并形成结合的无规共聚物(randomcopolymer)或者可根据聚合方法生成a、b规则性地排列而成的结构的嵌段共聚物(blockcopolymer)或a的聚合物中生成b的支链的接枝共聚物(graftedcopolymer)。
在上述具体例中,“共聚物(copolymer)”可以为无规共聚物、嵌段共聚物或接枝共聚物,例如,可以为无规共聚物,但并不一定不限定于此。
上述“亲水性单体(hydrophilicmonomer)”是指含有亲水性官能团的单体,具体地,可以为两性离子型单体、阴离子型单体、阳离子型单体或非离子型单体。更具体地,上述亲水性单体可以为亲水性烯属不饱和单体、2-甲基丙烯酰氧乙基磷酸胆碱(2-methacryloyloxyethylphosphorylcholine)及甜菜碱(betaine)中的一种或两种以上的混合物,但不限定于此。
上述亲水性烯属不饱和单体可包含丙烯酰胺、甲基丙烯酰胺、n-烷基丙烯酰胺、乙烯基磺酸、苯乙烯磺酸等,但不限定于此。
上述“2-甲基丙烯酰氧乙基磷酸胆碱(2-methacryloyloxyethylphosphorylcholine,mpc)”为具有两性离子(两性离子)的仿生单体且生物相容性非常优秀。因此,当使用2-甲基丙烯酰氧乙基磷酸胆碱来作为上述亲水性单体时,因生物适应性优秀而可安全地用于皮肤等人体中。
上述“甜菜碱(betaine)”是指一个分子中具有作为阳离子的季铵盐和酸(尤其为羧酸)的阴离子的分子,并且生物相容性非常优秀。
上述疏水性单体可以为丙烯酸脂的c6~c22烷基酯或甲基丙烯酸酯的c6~c22烷基酯,例如,为甲基丙烯酸酯的c18烷基酯,即,可以为甲基丙烯酸十八烷基酯(stearylmethacrylate),但不限定于此。
上述缔合型共聚物的亲水性单体及疏水性单体的含量比可以为1:0.0001~1,具体地,可以为1:0.0001~0.1,更具体地,可以为1:0.0001~0.01,进一步更具体地,可以为1:0.001~0.01,进一步更具体地,可以为1:0.005~0.01,进一步更具体地,可以为1:0.007~0.008,例如,可以为1:0.0075,但并不一定限定于此。
上述“表面改性”可以为指纤维素微丝的表面与缔合型共聚物相结合,例如,可以指缔合型共聚物接枝于纤维素微丝。
上述纤维素微丝的长度可以为10~5000nm,但不限定于此。
上述纤维素微丝的厚度可以为0.001~1000nm,但不限定于此。
本发明的再一具体例提供包含上述纤维素微丝的增稠剂组合物。
在本说明书中所使用的术语“增稠剂(thickener)”还可称为增稠剂、增稠稳定剂或稠化剂,是指使溶液之类的粘度增加的物质。
上述增稠剂组合物可以为使纤维素微丝分散于溶剂中的分散液,但并不一定限定于此,可使用本技术领域中周知的适当的方法来制备增稠剂组合物。
上述增稠剂组合物可具有1~1000000pa.s的粘度,具体地,可具有1~100000pa.s的粘度,但不限定于此。
存在于上述纤维素微丝的表面的缔合型共聚物可在水相中相互通过疏水性相互作用来形成纤维素凝胶网。该纤维素微丝凝胶相不仅表现出与现有增稠剂相似的性能,还在比较低剪切力条件下观察到剪切稀化(shearthinning)现象。当去除剪切力时,重新可逆地恢复初始粘度,由此可带来区别化的使用感。根据上述具体例的增稠剂组合物以纳米(nm)单位的网形成,因此可包含及渗透对皮肤有效的成分,但使皮肤外部的细菌或灰尘等的有害物质无法渗透,从而可保护皮肤。
本发明的还一具体例提供包含上述增稠剂组合物的化妆品组合物。
相对于100重量份的上述化妆品组合物,可包含0.001重量份至50重量份的上述增稠剂组合物,但并不一定限定于此,可根据化妆品组合物的剂型、用途等来适当选择量。
通过包含上述增稠剂组合物,上述化妆品组合物可具有保湿、特应性改善的效果。
根据上述具体例的化妆品组合物可包含脂肪物质、有机溶剂、增溶剂、胶凝剂、软化剂、抗氧化剂、悬浮剂、稳定剂、发泡剂(foamingagent)、芳香剂、表面活性剂、水、离子型或非离子型乳化剂、填充剂、多价螯合剂、螯合剂、防腐剂、维生素、阻隔剂、润湿剂、精油、染料、颜料、亲水性或亲油性活性剂、脂质囊泡或化妆品中通常所使用的任意其他成分等的化妆品学或皮肤科学领域中通常所使用的佐剂。上述佐剂以化妆品学或皮肤科学领域中通常所使用的量引入。
根据上述具体例的化妆品组合物的外型包含化妆品学或皮肤科学可接受的介质或基剂。它可以为适用于局部施用的所有剂型,能够以例如,溶液、凝胶、固体、糊状无水生成物、将油相分散于水相获得的乳液、悬浮液、微乳液、微胶囊、微颗粒球或离子型(脂质体)及非离子型的滤泡分散剂的形态提供,它们的组合物可根据该领域的常规方法来制备。
根据本发明一具体例的化妆品为选自由护肤乳液、皮肤柔软剂、皮肤调色剂、收敛剂、乳液、乳液、保湿乳液、营养乳液、按摩霜、营养霜、保湿霜、护手霜、粉底、精华液、营养精华液、面膜、肥皂、洁面泡沫、洁面乳、洁面霜、润肤露、沐浴露、洁面乳、护理液、美容液、美容面膜、软膏剂、凝胶剂、搽剂、液剂、贴剂及喷雾剂组成的组中的一种的剂型,但不限定于特定剂型,而是可具有常规的化妆品组合物的剂型。
本发明的另一具体例提供由缔合型共聚物表面改性而成的纤维素微丝的制备方法,上述方法可包括:步骤1),通过对包含纤维素、亲水性单体及疏水性单体的溶液进行活性自由基共聚反应来将缔合型共聚物接枝于纤维素表面;以及步骤2),通过添加2,2,6,6-四甲基哌啶-1-氧基(2,2,6,6-tetramethylpiperidine-1-oxyl)自由基引发剂来进行氧化反应并获得纤维素微丝。
上述“纤维素(cellulose)”为非常丰富的天然资源,可实现大量供给且具有优良的价格竞争力。并且,纤维素分子在结构上稳定,可呈现出耐化学性、耐盐性、耐酸碱性。
上述步骤1)的纤维素可以从木材、韩纸、纸、棉、麻、植物、海洋植物等中获得,但并不一定限定于此,可利用用于获得纤维素的常规方法来准备。
上述步骤1)的纤维素可以为在表面引入有活性自由基聚合反应的起始位点的纤维素。因此,上述步骤1)的纤维素可通过包括如下的步骤的方法来准备:步骤a),通过向纤维素中添加3-氨基丙基三乙氧基硅烷(3-aminopropyltriethoxysilane)来制备引入有胺基的纤维素;以及步骤b),向引入有胺基的纤维素中添加三氯乙酰基异氰酸酯(trichloroacetylisocyanate)来制备引入有三氯乙酰基的纤维素。
在上述步骤b)中所引入的三氯乙酰基可作为活性自由基聚合反应的起始位点。
相对于溶液总重量,上述步骤1)的纤维素可包含1~15重量百分比,具体地,可包含1~10重量百分比,例如,可包含3~5重量百分比,但不限定于此。
上述步骤1)的亲水性单体可以为两性离子型单体、阴离子型单体、阳离子型单体或非离子型单体。
更具体地,上述亲水性单体可以为亲水性烯属不饱和单体、2-甲基丙烯酰氧乙基磷酸胆碱(2-methacryloyloxyethylphosphorylcholine)及甜菜碱(betaine)中的一种或两种以上的混合物,但不限定于此。
上述亲水性烯属不饱和单体可包含丙烯酰胺、甲基丙烯酰胺、n-烷基丙烯酰胺、乙烯基磺酸、苯乙烯磺酸等,但不限定于此。
上述步骤1)的疏水性单体可以为丙烯酸脂的c6~c22烷基酯或甲基丙烯酸酯的c6~c22烷基酯的纤维素微丝,例如,甲基丙烯酸酯的c18烷基酯,即,可以为甲基丙烯酸十八烷基酯(stearylmethacrylate),但不限定于此。
上述步骤1)的溶液还可包含能够用于活性自由基聚合反应的催化剂,例如,还可包含羰基钼(mo(co)6),但不限定于此。
上述步骤1)的缔合型共聚物的亲水性单体及疏水性单体的含量比可以为1:0.0001~1,具体地,可以为1:0.0001~0.1,更具体地,可以为1:0.0001~0.01,进一步更具体地,可以为1:0.001~0.01,进一步更具体地,可以为1:0.005~0.01,进一步更具体地,可以为1:0.007~0.008,例如,可以为1:0.0075,但并不一定限定于此。
在未做其他处理的情况下,纤维素以纤维束形成,当通过进行上述步骤2)来将纤维素纤维束转换为具有数纳米的厚度和数微米大小的长轴的微丝时,从水相形成透明的凝胶相,并可表现出增稠效果。由此制备的纤维素微丝因强大的水合行为而表现出优秀的增稠效果,并且因纤维之间的物理相互作用而呈现出区别化的流变行为。
具体实施例
以下,在本发明的实施例中进一步详细说明本发明。然而,以下实施例仅为用于例示本发明的内容,并不是用于限定或限制本发明的发明要求保护范围。本发明所属技术领域的普通技术人员可从本发明的详细说明及实施例容易地类推出,这可解释为属于本发明的发明要求保护范围。
<实施例1>:由缔合型共聚物表面改性而成的纤维素微丝的制备
以下,通过实施例1-1至1-4的方法来制备了由缔合型共聚物表面改性而成的纤维素微丝。整体合成过程在图1中示出。
1-1:向纤维素表面引入胺基
在甲苯中固定具有多种长度及厚度的纤维素3wt%并进行了分散。接着,为了引入胺基,添加相对于纤维素的重量的150%的3-氨基丙基三乙氧基硅烷(3-aminopropyltriethoxysilane),并在110℃温度条件下与纤维素表面的羟基(-oh)反应了8小时。通过反复的离心分离来清洗及回收引入了胺基(-nh2)的纤维素。
1-2:向纤维素表面引入三氯乙酰基
为了在纤维素中引入活性自由基聚合反应(livingradicalpolymerization)的起始位点,进行了纤维素粒子的表面中所引入的胺基和三氯乙酰基异氰酸酯(trichloroacetylisocyanate)的缩合反应。为此,在35℃温度条件下,在甲苯中将含有在上述实施例1-1中所制备的胺基的纤维素粉末照射超声波10分钟并进行了再分散。此时,纤维素的浓度被固定为3wt%。使相对于分散的纤维素的重量的50%的三氯乙酰基异氰酸酯(trichloroacetylisocyanate)与二月桂酸二丁基锡(dibutyltindilaurate)催化剂一同在氩气氛下的80℃温度条件下搅拌8小时,从而合成了在表面引入了三氯乙酰(trichloroacetyl)基的纤维素。通过反复的离心分离来清洗及回收了引入了三氯乙酰基(-coccl3)的纤维素。
1-3:利用表面聚合的缔合型聚合物链的引入
在乙醇中,将在实施例1-2中所制备的在表面保留有聚合起始位点的纤维素粉末在35℃温度条件下照射10分钟超声波并进行了再分散。此时,纤维素的浓度被固定为3wt%。接着,将作为仿生单体的2-甲基丙烯酰氧乙基磷酸胆碱(2-methacryloyloxyethylphosphorylcholine,mpc)和保留有c18疏水性链的甲基丙烯酸十八烷基酯(stearylmethacrylate,sma)投入于上述分散液中。催化剂使用了羰基钼(mo(co)6),并且相对于总重量投入了0.05wt%。向反应器中注入氩来去除氧气之后,在70℃温度条件下,进行12小时的聚合物聚合反应(活性自由基聚合反应),最终合成了在表面引入了双性离子聚合物(zwitterionicpolymer)和疏水性链的纤维素。其中,2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)以无规聚合的方式引入纤维素表面,因此,合成后,在主链中2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)无规则排列。
1-4:通过氧化反应的纤维素微丝分散液的获得
将在实施例1-3中所制备的在表面聚合有聚合物的纤维素在常温下通过搅拌再分散于水中。此时,纤维素的浓度被固定为1wt%。将相对于分散的纤维素的重量的10-30%的nabr与2,2,6,6-四甲基哌啶-1-氧基(2,2,6,6-tetramethylpiperydine-1-oxylradical)催化剂一同投入于分散液中,并在常温条件下搅拌了10分钟。接着,添加1~15mmol的naclo,并利用0.5m的naoh来滴定,使得反应溶液的ph为10,然后在常温下以600rpm搅拌了4小时。通过该氧化反应,纤维素表面的残留的羟基经由醛氧化为羧酸,纤维素纤维束展开为微纤维单位并获得了纤维素微丝。
<比较例1>:未进行表面改性的纤维素微丝分散液的制备
为了比较向纤维素表面引入的双性离子聚合物和疏水性链的效果,仅利用纤维素制备了分散液。
具体地,通过将纤维素分散于水中来制备了未进行表面改性的纤维素微丝分散液。
<实验例1>:确认是否引入缔合型聚合物
对在实施例1中所制备的利用缔合型聚合物进行表面改性的纤维素微丝和在比较例1中所制备的未进行表面改性的纤维素微丝进行了用于确认是否引入缔合型聚合物的实验。
利用透射电子显微镜(transmissionelectronmicroscope,tem)分析确认了纤维素微丝表面中的作为缔合型聚合物的poly(mpc-co-sma)化学合成的纤维的形态(图2)。
并且,为了验证2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)的合成效率,进行了氢核磁共振(1h-nmr)分析。具体地,分析了在实施例1中进行表面聚合时存在于溶剂中的单体的转化率。其结果,利用2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)的表面改性的纤维素微丝的合成效率呈现出70%(图3)。
并且,作为利用傅里叶变换红外光谱学(ft-ir,fouriertransforminfraredspectrophotometer)的定性分析结果,确认了实施例1中所制备的纤维素微丝表面中引入了2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)(图4)。
<实验例2>:纤维素微丝中的根据甲基丙烯酸十八酯(sma)的比率的粘度确认实验
进行了比较确认根据引入于纤维素表面的甲基丙烯酸十八酯(sma)的比率的粘度增加程度的实验。
具体地,纤维素微丝的重量比设定为4wt%,除了对各个实验的变数值以外,均以相同方式进行。除了在实施例1-3的步骤中设定了不同的2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma)的比率之外,均已与实施例1相同的方法进行了由缔合型共聚物表面改性而成的纤维素微丝的制备。φsma是指对于2-甲基丙烯酰氧乙基磷酸胆碱(mpc)的甲基丙烯酸十八酯(sma)的相对含量比率。即,表面改性的纤维素微丝对不利用甲基丙烯酸十八酯(sma)而仅利用2-甲基丙烯酰氧乙基磷酸胆碱(mpc)来制备的实验组(φsma=0)、利用mpc:sma=1:0.005的比率来制备的实验组(φsma=0.005)、利用mpc:sma=1:0.0075的比率来制备的实验组(φsma=0.0075)、利用mpc:sma=1:0.01的比率来制备的实验组(φsma=0.01),测定了粘度,并使用比较例1的未进行表面改性的纤维素微丝(neatcnfs)作为对照组。粘度利用了美国ta公司的流变仪(型号:dhr1)并通过将根据剪切速度的剪切应力换算为粘度的方法进行了测定。对此的实验结果分别在图5中列出。
如在图5中所确认,通过最佳的甲基丙烯酸十八酯(sma)(φsma=0.0075)所共聚的疏水性缔合型链的引入,相对于对照组,增加了约100倍的粘度。并且,在低剪切速度条件下,纤维素形成凝胶相并呈现出高粘度。但是,随着剪切速度的增加,凝胶相击垮并确认到了粘度急剧降低的现象。疏水性相互作用根据有无剪切速度来表现出,因此可确认相对于剪切速度的可逆性的粘度变化。
<实验例3>:利用流变仪的缔合型纤维素微丝分散液的流变的确认
按照各个甲基丙烯酸十八酯(sma)含量的效果的流变的确认以如下方法进行。分别比较了根据甲基丙烯酸十八酯(sma)含量的模量效果、耐盐性效果、耐酸碱性并进行了确认。
纤维素微丝重量比设定为4wt%,除了对各个实验的变数值以外,均以相同的方法进行。分别使用了mpc:sma=1:0、1:0.005、1:0.0075或1:0.01比率并以与实施例1相同的方法来制备了表面改性的纤维素微丝。使用根据比较例1制备的未进行表面改性的纤维素微丝(neatcnfs)来作为对照组。弹性模量(储能模量,storagemodulus)为利用将根据剪切变形率(strain(%))的剪切应力换算为弹性模量的方法来进行了测定。粘性模量(损耗模量,lossmodulus)为利用将根据剪切变形率(strain(%))的剪切应力换算为粘性模量的方法进行了测定。剪应力(shearstress)为测定了根据剪切速度及剪切变形率所取得的值。屈服应力为利用换算弹性模量及粘性模量的方法来进行了测定。所测定的弹性模量在图6a中示出,粘性模量在图6b中示出,剪切应力在图6c中示出,屈服应力在图6d中示出。
其结果,可确认出本发明的纤维素微丝具有优秀的弹性力和粘度,尤其,以mpc:sma=1:0.0075含量比制备的表面改性的纤维素微丝的效果最为优秀(图6a至图6d)。
以如下方法进行了耐盐性及耐酸碱性的测定实验。利用mpc:sma=1:0.0075的比率以与实施例1相同的方法制备了表面改性的纤维素微丝(associativecellulosenanofibers,acnfs)。使用根据比较例1制备的未进行表面改性的纤维素微丝(neatcnfs)来作为对照组。为了确认耐盐性,相对于各个微丝分散液,添加了0、1或1.5m的nacl之后,以与实验例2相同的方法测定了根据剪切速度的粘度。并且,为了确认耐酸碱性,将各个微丝分散液调节为ph2、7或11之后,以与实验例2相同的方法测定了根据剪切速度的粘度。耐盐性测定结果在图7a中示出,耐酸碱性测定结果在图7b中示出。
其结果,本发明的纤维素微丝呈现出优秀的耐盐性和耐酸碱性(图7)。基于疏水性相互作用来缔合的纤维素微丝凝胶相可解释为只要不受到离子浓度的变化就可保持固有的凝胶相。
从以上结果确认了纤维素中成功引入了2-甲基丙烯酰氧乙基磷酸胆碱(mpc)和甲基丙烯酸十八酯(sma),并流变学上验证了通过sma的增稠和粘度稳定性效果。
工业实用性
利用本发明的纤维素微丝的增稠剂组合物可广泛适用于化妆品组合物、皮肤外用药组合物等化妆品市场,并预期在基于复合流体的多种工业中的利用度相当高。
- 还没有人留言评论。精彩留言会获得点赞!