苯并咪唑衍生物的酸加成盐的制作方法
本发明涉及具有优异的稳定性及溶解性的苯并咪唑衍生物的酸加成盐以及其制备方法。
背景技术:
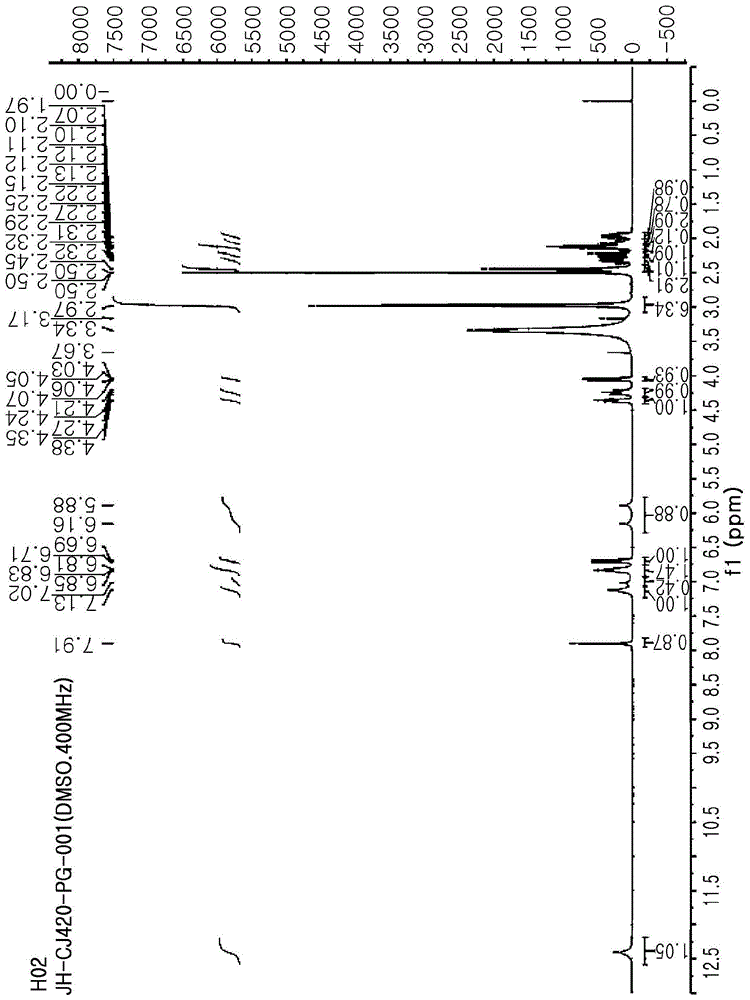
:由下式1表示的4-[((4s)-5,7-二氟-3,4-二氢-2h-色烯-4-基)氧基]-n,n,2-三甲基-1h-苯并咪唑-6-甲酰胺[(s)-4-((5,7-二氟色满-4-基)氧基]-n,n,2-三甲基-1h-苯并[d]咪唑-6-甲酰胺]是分子量为387.39的药物活性成分:[式1]此化合物是用于预防及治疗由酸泵拮抗活性介导的疾病的药物成分,所述疾病包括(但不限于)胃肠疾病,例如胃食道疾病、胃食道逆流病(gerd)、消化性溃疡、胃溃疡及十二指肠溃疡、由nsaid诱导的溃疡、胃炎、幽门螺旋杆菌感染、消化不良及功能性消化不良、卓-艾综合征(zollinger-ellisonsyndrome)、非糜烂性逆流病(nerd)、内脏痛、胃灼热、恶心、食道炎、吞咽困难、流涎、气道病变及哮喘。国际专利第wo2007072146号公开了用色满取代的苯并咪唑衍生物,其用作酸泵抑制剂(h+k+atp酶抑制剂)。国际专利第wo2016117814号也公开了上文所提及的式1的新晶形及其制备方法。具体地,该专利公开了上文所提及的式1化合物的结晶游离碱,其有利于制备制剂,原因在于:在长期及应力储存条件下稳定,未观察到因随时间推移的变化所致的晶体转变,具有优异的光稳定性且具有低吸湿性及低静电感应。上文所提及的化合物对盐的选择有限,原因在于该化合物具有非常低的水溶性(0.02mg/ml,ph6.8),在酸性条件下溶解度增加(0.7mg/ml,ph3.0),但效果不大,且在酸性条件下降解产物增加,使得该化合物不具有良好稳定性。此外,在使用增溶剂(例如表面活性剂)以改善溶解度的情形下,需要过量的赋形剂,因此产生困难。同时,就制剂而言,游离碱或结晶酸加成盐不能确保显著的水溶性。具体地,当考虑注射制剂时,除溶解速率以外需要确保沉淀稳定性,使得在血浆ph下其沉淀不会发生。因此,充分确保动力学及热力学溶解度二者是重要的。然而,非晶酸加成盐由于热力学溶解度低且在酸性条件下同时降解的化合物特性,非常难以同时确保溶解性及稳定性。就药物而言,多形晶形是重要的,此乃因多形晶形可根据药物以各种形式存在,且特定多形晶形可影响制备药物成分的容易性、溶解度、储存稳定性、制备完整药物产品的容易性及体内药物活性。选择优异晶形的标准基于药物所需的最重要的物理化学特性,且优化晶形的选择可根据目的而变化,该目的为例如选择热力学上最稳定的晶形、对于药物成分及完整药物产品选择药物上优化的晶形、改善药物的溶解度及溶解速率或改变药物动力学特性。同时,在非晶形药物成分的情形下,颗粒的表面积增加,因此动力学溶解度通常增加,但在体内ph条件下改善热力学溶解度仍存在限制。此外,非晶形药物成分不具有由结晶所引起的晶格能,不具有明显熔点,且与相同的结晶药物成分相比具有显著低的稳定性。同时,当在药物上制备注射制剂时,非晶形药物成分通常作为能够制备为冻干材料的固体使用。然而,在此情形下,非晶形药物成分必须确保其制剂内的稳定性以及同时确保溶解度及沉淀稳定性。因此,本发明人努力开发能够用作热力学上非常稳定的注射制剂的药物成分的酸加成盐,而由上文所提及的式1所表示的化合物同时具有优异的水溶性及沉淀稳定性。结果,经鉴别,由式1所表示的化合物的吡酮酸盐及苹果酸盐同时具有优异的稳定性及溶解度,由此完成本发明。现有技术参考文献专利文件国际专利第wo2007072146号国际专利第wo2016117814号技术实现要素:技术问题本发明的目标是提供由下式1表示的化合物的吡酮酸盐或苹果酸盐以及其制备方法。[式1]技术方案在解决上文所提及任务的一个方面,本发明提供由下式1表示的化合物的吡酮酸盐或苹果酸盐。[式1]具体而言,式1化合物的吡酮酸盐可以是由下式2表示的吡酮酸盐,且式1化合物的苹果酸盐可以是由下式3表示的苹果酸盐。[式2][式3]在本发明中,根据钾竞争性酸阻滞剂(p-cab)的药理学机制,式1化合物是用于预防及治疗胃肠疾病及与其相关的出血的新材料。式1化合物在ph6.8的缓冲溶液中的溶解度仅为0.02mg/ml,因此在活体内ph条件下具有非常低的水溶解度。另外,下式4的降解产物在溶解度增加的酸性环境中继续随暴露时间成比例地增加。[式4]具体而言,式4的降解产物在酸性条件下通过下式5的机制断开醚键所产生,且随着ph变低或在低ph中暴露时间变长,降解产物趋于成比例地增加。[式5]式1化合物在ph3的缓冲溶液条件下的水溶解度仅为0.7mg/ml,使得需要更低的ph来增加溶解度。然而,在此情形下,维持稳定性存在困难。另外,即使溶解度在低ph中得以改善,但在活体内ph条件下未必发生沉淀,因此如本发明中所要求的式1化合物的物理性质必须同时满足以下四个条件:1)当考虑剂量及体积时,优选确保溶解度为50mg/ml或更高;2)在其至少10mg/ml溶解于ph6.8的缓冲溶液中的状态下,即使随着时间推移也无降解产物沉淀;3)确保足够的稳定性以用作药物成分;及4)在储存期间维持非晶形态且不因随时间推移的变化而转化为结晶。本发明人意在制备式1化合物与水溶性氨基酸之间的共晶体以满足上文所提及的四个条件。就此而言,欧洲专利公开第ep2493457号公开了一种固体复合物,其以使得氨基酸与药物结合为基质形成剂的方式快速溶解。因此,本发明的式1化合物不得在快速溶析的同时在活体内ph条件下沉淀。然而,在如酸加成盐通过降低ph而导致快速溶析的情形下,在活体内ph环境(接近中性)中离子化程度下降,使得可存在诱发固体沉淀的可能性。因此,本发明人努力通过分子间键结以及水溶性氨基酸与式1化合物之间的氢键的介质来制备共晶体,以便相对较少地受ph影响。下表1显示在本发明中用作共晶体形成剂的氨基酸以及能够形成共晶体的化合物,且将共晶体形成剂的当量设定为与式1化合物相比的1:1摩尔比。[表1]下表2指示当以1:1摩尔比在表1的共晶体形成剂与式1化合物之间形成共晶体时在室温下的可结晶性及水溶解度。具体而言,表1中的1号至6号共晶体形成剂形成结晶共晶体,且7号至12号共晶体形成剂显示非结晶特性,其中其结果通过pxrd来鉴别。同时,作为结晶共晶体的实例,式1化合物与l-丙氨酸之间的共晶体能够通过如下式6中所示的氢键介质来指示结晶特性。[式6][表2]基于表2的结果,经鉴别,本发明中的式1化合物在与由一组具有水溶性氨基酸及氢键供体及受体的相关化合物组成的共晶体形成剂反应结合后,水溶解度改善不大。然而,经鉴别,与结晶固体相比,非结晶固体具有相对较高的溶解度改善程度,使得本发明基于上文所提及的结果而完成。根据本发明,满足所有上文所提及条件的盐为式1化合物的吡酮酸盐及式1化合物的苹果酸盐。具体而言,式1化合物的吡酮酸盐(由式2表示)是以式1化合物与焦谷氨酸形成盐的方式形成的,且焦谷氨酸能够用于具有与焦谷氨酸盐、吡酮酸或吡酮酸盐相同的含义。具体而言,焦谷氨酸可以是l-焦谷氨酸。同样地,式1化合物的苹果酸盐(由式3表示)是以式1化合物与苹果酸形成盐的方式形成的,且苹果酸能够用于具有与苹果酸盐相同的含义。具体而言,苹果酸可以是l-苹果酸。式1化合物的吡酮酸盐或式1化合物的苹果酸盐可优选为非结晶或部分结晶,且最优选为部分结晶,其中基于盐的总wt%,至少95wt%为非结晶。在本发明的一个例示性实施方式中,作为比较式1化合物的12种的非晶形酸加成盐的溶解度结果,与其他盐相比,富马酸盐、草酸盐、柠檬酸盐、l-焦谷氨酸盐、l-苹果酸盐及l-酒石酸盐显示更优异的溶解度(表4)。在该六种盐中,经鉴别,与其他盐相比,l-焦谷氨酸盐及l-苹果酸盐显示更优异的液相稳定性及固相稳定性(表5及6),且甚至在活体内ph6.8条件下维持均质溶液(表7),因此可看出,式1化合物的吡酮酸盐及式1化合物的苹果酸盐是式1化合物优化制剂的药物成分。在另一方面,本发明提供用于预防或治疗由酸泵拮抗活性所介导的疾病的药物组合物,其包括式1化合物的吡酮酸盐或苹果酸盐。式1化合物的吡酮酸盐及式1化合物的苹果酸盐例如为上文所阐述的盐。由酸泵拮抗活性所介导的疾病可以是选自由以下组成的组中的疾病:胃食道疾病、胃食道逆流病(gerd)、消化性溃疡、胃溃疡及十二指肠溃疡、由nsaid诱导的溃疡、胃炎、幽门螺旋杆菌感染、消化不良及功能性消化不良、卓-艾综合征、非糜烂性逆流病(nerd)、内脏痛、胃灼热、恶心、食道炎、吞咽困难、流涎、气道病变及哮喘。本发明的药物组合物可以是一种选自由以下组成的组的制剂:粉末、颗粒、锭剂、胶囊、悬浮液、乳液、糖浆、气溶胶、软膏剂、霜剂、栓剂及注射剂,但并不限于此。具体而言,药物组合物可由于本发明的药物组合物中含有的式1化合物的吡酮酸盐或苹果酸盐(由式2或3表示)的特性而为注射制剂,此乃因吡酮酸盐或苹果酸盐在活体内ph环境中显示优异的稳定性。含于本发明的组合物中的活性组分的量根据欲给药的患者的状态、目标治疗程度等而变化。优选地,本发明的组合物可以以1至100mg/ml、优选1至50mg/ml的浓度含有作为由式2表示的化合物或由式3表示的化合物中的活性组分存在的式1化合物。若活性组分以1mg/ml或更低的低浓度含有,则可给予大剂量的注射剂以展现足够的治疗效应,因此对给予患者的受影响部位造成困难。另一方面,若以100mg/ml或更高的高浓度含有活性组分,则可能难以符合在再悬浮或溶解时可出现杂质或可产生沉淀的同时甚至溶解稳定剂的组合物。当制备根据本发明的注射组合物时,可使用注射用水来制备注射组合物。根据本发明,含有式1化合物的药物上可接受的盐的注射剂可选择性地包含等渗剂、缓冲溶液、渗透剂等,这些试剂在本领域内习用,但并不限于此。同样,本发明提供由式1表示的化合物的吡酮酸盐或苹果酸盐的用途,以用于预防或治疗由酸泵拮抗活性所介导的疾病。此外,本发明提供由式1表示的化合物的吡酮酸盐或苹果酸盐的用途,以用于制备用于预防或治疗由酸泵拮抗活性所介导的疾病的药物。用于制备药物的由式1表示的化合物所表示的吡酮酸盐或苹果酸盐可与可接受的佐剂、稀释剂、载体等混合,且可连同其他活性剂制备为复合制剂,由此对活性组分产生协同效应。此外,本发明提供预防或治疗由酸泵拮抗活性所介导的疾病的方法,其包含:给予治疗有效量的由式1表示的化合物的吡酮酸盐或苹果酸盐。根据本发明,上文所提及的“对象”包含哺乳动物,特别地,人类。本发明中所使用的术语“治疗有效量”意指盐的预防或治疗由酸泵拮抗活性所介导的疾病的这一有效量。根据本发明,预防或治疗由酸泵拮抗活性所介导的疾病的方法包含:给予上文所提及的盐,由此在其症状表现之前解决疾病自身以及抑制或避免此等症状。在管控疾病方面,特定活性组分的预防或治疗剂量根据疾病的性质及严重程度或其状态以及活性组分的给予途径而有所变化。剂量及其频率根据个别患者的年龄、重量及反应而有所变化。当然,考虑这些因素的本领域技术人员可容易地选择适当投用方案。除非其彼此矛盾,否则本发明的使用、组合物及治疗方法中所提及的事项相等地起作用。在另一方面,本发明提供一种制备式1所表示化合物的焦谷氨酸盐或苹果酸盐的方法,其中该方法包含:a)将式1所表示的化合物及焦谷氨酸或苹果酸作为酸加成盐溶解于有机溶剂中;b)在减压下浓缩步骤a)中所制备的溶液以沉淀固体,然后向其添加共溶剂以搅拌所得溶液;及c)过滤出并干燥沉淀的固体。更具体而言,本发明的制备方法包含:步骤a)将式1所表示的化合物溶解于有机溶剂中并于其中溶解酸加成盐。此时,上文所使用的酸为焦谷氨酸或苹果酸。若使用其他酸来制备酸加成盐并将其溶解,则所制备的式1化合物的盐具有低液相或固相稳定性,例如未改善溶解度或产生降解产物或杂质,且可能引起问题,例如沉淀固体。具体而言,焦谷氨酸可以是l-焦谷氨酸,且苹果酸可以是l-苹果酸。有机溶剂可以是甲醇,但并不限于此,其中与式1所表示的化合物相比,有机溶剂可以以5至20倍(体积/重量)、优选10倍(体积/重量)添加。若有机溶剂的添加量比式1所表示的化合物的5倍少或比20倍多,则所制备的盐中所含吡酮酸盐或苹果酸盐的量可能不适合展现优异的溶解性及稳定性。更具体而言,本发明的制备方法包含:在减压下浓缩步骤a)中所制备的溶液以沉淀固体,然后向其添加共溶剂以搅拌所得溶液。共溶剂可以是丙酮及乙酸乙酯的混合溶剂,但并不限于此,其中与式1所表示的化合物相比,共溶剂可以以1至10倍(体积/重量)、优选5倍(体积/重量)添加。若有机溶剂的添加量比式1所表示的化合物的1倍少或比10倍多,则所制备的盐中所含的吡酮酸盐或苹果酸盐的量可能不适合展现优异的溶解性及稳定性。丙酮与乙酸乙酯之间的共溶剂的比率可以是丙酮:乙酸乙酯=1:4(v/v),但并不限于此。更具体而言,本发明的制备方法包含:过滤出并干燥步骤b)中所沉淀的固体。过滤可以是减压过滤,但并不限于此。干燥可通过常规干燥方法来实施,例如冷冻干燥、旋转蒸发干燥、喷雾干燥、真空干燥或流化床干燥,特别地,真空干燥。。有益效果本发明中所提供的式1化合物的新非晶形酸加成盐由于同时具有优异的物理化学特性、稳定性及溶解度,因此能够容易地用作用于口服制剂及注射制剂的药物成分。附图说明图1显示根据本发明的实施方式制备的式1的非晶形化合物的吡酮酸盐的1h-nmr结果。图2显示根据本发明的实施方式制备的式1的非晶形化合物的苹果酸盐的1h-nmr结果。图3显示根据本发明的实施方式制备的式1的非晶形化合物的吡酮酸盐的pxrd结果。图4显示根据本发明的实施方式制备的式1的非晶形化合物的苹果酸盐的pxrd结果。图5显示根据本发明的实施方式制备的式1的非晶形化合物的富马酸盐的pxrd结果。图6显示根据本发明的实施方式制备的式1的非晶形化合物的草酸盐的pxrd结果。图7显示根据本发明的实施方式制备的式1的非晶形化合物的柠檬酸盐的pxrd结果。图8显示根据本发明的实施方式制备的式1的非晶形化合物的酒石酸盐的pxrd结果。图9显示式1的结晶化合物的游离碱的dsc结果。图10显示根据本发明的实施方式制备的式1的非晶形化合物的吡酮酸盐的dsc结果。图11显示根据本发明的实施方式制备的式1的非晶形化合物的苹果酸盐的dsc结果。具体实施方式在下文中,将根据以下实施例更详细地阐述本发明的构成和效果,这些实施例出于说明而提出,但不应解释为限制本发明。实施例1:制备式1化合物的吡酮酸盐在25℃下将100g式1的结晶化合物及34.98gl-焦谷氨酸(1.05eq.)完全溶解于1000ml甲醇中,然后将所得溶液在50℃下浓缩,同时在减压下搅拌直至固体沉淀为止。在25℃下将丙酮及乙酸乙酯的共溶剂以丙酮:乙酸乙酯=1:4(500ml)的比率添加至浓缩物,并将所得溶液剧烈搅拌30分钟。将固体在减压下过滤出,利用100ml乙酸乙酯洗涤,并在40℃下真空干燥16小时,使得获得113.8g(产率85.4%)呈白色粉末形式的式1的非晶形化合物的吡酮酸盐(图1)。实施例2:制备式1化合物的苹果酸盐在25℃下将100g式1的结晶化合物及36.33gl-苹果酸(1.05eq.)完全溶解于1000ml甲醇中,然后将所得溶液在50℃下浓缩,同时在减压下搅拌直至固体沉淀为止。在25℃下将丙酮及乙酸乙酯的共溶剂以丙酮:乙酸乙酯=1:4(500ml)的比率添加至浓缩物,并将所得溶液剧烈搅拌30分钟。将固体在减压下过滤出,利用100ml乙酸乙酯洗涤,并在40℃下真空干燥16小时,使得获得120.4g(产率89.4%)呈白色粉末形式的式1的非晶形化合物的苹果酸盐(图2)。比较例1:制备式1化合物的非晶形游离碱在25℃下将100g式1化合物完全溶解于1000ml甲醇中,然后将所得溶液在50℃下浓缩,同时在减压下搅拌直至固体沉淀为止。在25℃下将丙酮及乙酸乙酯的共溶剂以丙酮:乙酸乙酯=1:4(500ml)的比率添加至浓缩物,并将所得溶液剧烈搅拌30分钟。将固体在减压下过滤出,利用100ml乙酸乙酯洗涤,并在40℃下真空干燥16小时,使得获得94.7g(产率94.7%)呈白色粉末形式的式1的非晶形化合物的游离碱。实验例1:关于溶解度的比较测试如表3中所示选择12种酸后制备非晶形酸加成盐,使得在通过酸加成盐使化合物质子化/酸化后,阴离子不能作为亲核剂,同时增强式1化合物的溶解度。[表3]具体而言,在不包括1.5-萘二磺酸的11种酸的情形下,酸加成盐以使得其当量为每种酸与式1化合物之间的摩尔比为1:1的方式来制备。在1.5-萘二磺酸的情形下,半盐以使得其当量为此酸与式1化合物之间的摩尔比为0.5:1的方式来制备。上文所提及的制备方法与实施例1及2中相同。下表4指示12种非晶形酸加成盐在室温下的水溶解度。每一水溶解度通过过饱和方法来量测。每一溶解度并非基于式1的游离碱而基于所量测的酸加成盐本身的溶解度转化,其中其结果指示于下表4中。也量测比较例1中式1化合物的非晶形游离碱的溶解度,其中其结果指示于下表4中。[表4]如表4中所见,经鉴别,在12种酸加成盐中,6种类型的盐指示水溶解度为50mg/ml或更高。具体而言,富马酸盐、草酸盐、柠檬酸盐、l-焦谷氨酸盐、l-苹果酸盐及l-酒石酸盐展现对改善溶解度的效应。特别地,草酸盐、l-焦谷氨酸盐及l-苹果酸盐也经鉴别展现100mg/ml或更高的显著优异的溶解度。基于上文所提及的结果可看出,与非晶形游离碱相比,式1化合物的非晶形酸加成盐具有极大增加的溶解度,尤其根据盐类型而显示对改善溶解度的不同效果。实验例2:x-射线粉末绕射(pxrd)分析实施pxrd分析以分析存在于式1化合物的盐中的式1化合物的结晶变化。具体而言,将如上文所提及的实验例1中所鉴别的水溶解度为50mg/ml或更高的式1化合物的六种类型的非晶形酸加成盐(l-焦谷氨酸盐、富马酸盐、草酸盐、柠檬酸盐、l-苹果酸盐及l-酒石酸盐)分别插入每一pe袋中,然后将所得的袋储存在40℃及75%rh条件下的稳定室内一个月。然后,在x射线粉末衍射仪(d8advance)上使用cu-kα线来实施盐的pxrd分析。衍射仪配备有渗透,且将电流容量设定为45kv及40ma。将发散狭缝及散射狭缝设定为1°且将接收狭缝设定为0.2mm。自5至35°2θ使用3°/分钟(0.4秒/0.02°间隔)的θ-2θ连续扫描。作为储存后一个月检查pxrd的结果,经鉴别,所有类型的盐均维持非晶形,因此未观察到晶体转变(图3至8)。实验例3:关于液相稳定性的比较测试将如实验例1中所鉴别的水溶解度为50mg/ml或更高的式1化合物的六种类型的非晶形酸加成盐(l-焦谷氨酸盐、富马酸盐、草酸盐、柠檬酸盐、l-苹果酸盐及l-酒石酸盐)分别溶解于纯化水中以制备成浓度为20mg/ml,然后将所得溶液在40℃下搅拌,使得溶液在初始阶段及第一天用hplc分析。因此,将降解产物(式4化合物)的产生量转化成与呈无多形性的稀释状态的式1化合物的曲线下面积(auc)相比的百分比(%),其中其结果指示于下表5中。[表5]结果,经鉴别,式1化合物的l-焦谷氨酸盐及l-苹果酸盐的液相稳定性较其他盐优异至少三倍,且由式4化合物所表示的降解产物在式1化合物的草酸盐中产生最多。因此,鉴于其中降解产物必须管控为0.1%或更低的关于注射制剂的活性药物成分(api)的ich指南,经鉴别,仅l-焦谷氨酸盐及l-苹果酸盐为药物上可用且稳定的酸加成盐。同时,就物理性质而言,经鉴别,柠檬酸盐及富马酸盐形成黏性凝胶而不维持均质液相状态,而其余四种盐维持均质液相状态。实验例4:关于固相稳定性的比较测试将如上文所提及的实验1中所鉴别的水溶解度为50mg/ml或更高的式1化合物的六种类型的非晶形酸加成盐(l-焦谷氨酸盐、富马酸盐、草酸盐、柠檬酸盐、l-苹果酸盐及l-酒石酸盐)分别插入每一pe袋中,然后将所得袋储存在40℃及75%rh条件下的稳定室内一个月。然后,将通过karlfischer分析检查的水的产生量、通过hplc分析检查的杂质的产生量及在与实验例3相同的条件下量测的降解产物(式4化合物)的产生量分别转化成面积百分比,其中其结果指示于下表6中。[表6]结果,与液相稳定性的结果类似,在式1化合物的l-焦谷氨酸盐及l-苹果酸盐中固相加速稳定性最优异,且杂质总量在富马酸盐中最大。因此,鉴于其中杂质必须管控为0.1%或更低的关于注射制剂的活性药物成分(api)的ich指南,经鉴别,仅l-焦谷氨酸及l-苹果酸盐为药物可用且稳定的酸加成盐。同时,就物理性质而言,在所有类型的盐中均未观察到吸湿性,且经肉眼鉴别其中维持白色粉末类型。实验例5:关于沉淀稳定性的比较测试将如实验例1中所鉴别的水溶解度为50mg/ml或更高的式1化合物的六种类型的非晶形酸加成盐(l-焦谷氨酸盐、富马酸盐、草酸盐、柠檬酸盐、l-苹果酸盐及l-酒石酸盐)分别以10mg/ml的浓度完全溶解于ph6.8的缓冲溶液中,并在37℃下储存24小时。然后,观察此等溶液的状态,其中其结果指示于下表7中。[表7]酸加成盐土壤沉淀或不沉淀富马酸沉淀草酸沉淀柠檬酸沉淀l-焦谷氨酸维持均质溶液l-苹果酸维持均质溶液l-酒石酸维持均质溶液结果,经鉴别,在富马酸盐、草酸盐及柠檬酸盐中有固体沉淀,而在l-焦谷氨酸盐、l-苹果酸盐及l-酒石酸盐中维持均质溶液。因此,经鉴别,l-焦谷氨酸盐、l-苹果酸盐及l-酒石酸盐具有优异的沉淀稳定性。实验例6:温度差示扫描量热法(dsc)分析使用可自tacompany获得的dscq20在氮纯化下以10℃/min的扫描速度自20至300℃在封闭风扇中实施dsc量测。具体而言,对式1的结晶化合物(其为实施例1及2的反应物)以及非晶形吡酮酸盐及苹果酸盐(其为产物)实施dsc量测。结果,经鉴别,非晶形酸加成盐(吡酮酸盐及苹果酸盐)自式1的结晶化合物的游离碱合成(图9至11)。根据上文所提及的结果,经鉴别,式1化合物的吡酮酸盐(由式2表示)以及式1化合物的苹果酸盐(由式3表示)为用于注射制剂的药物成分,其针对式1的苯并咪唑衍生物优化。尽管本发明的特定部分已于上文中详细地阐述,但本领域技术人员应明了,这些详细阐述仅为例示性实施方式,而不应解释为限制本发明的范围。因此,本发明的实质范围将基于随附权利要求及其等效形式来界定。本文中省略了关于本领域技术人员可容易理解并推断的内容的详细阐述。除本说明书中所阐述的详细实施例以外,可在不背离本发明的技术理念或本质特征的情形下进行各种修改。因此,本发明可以除本说明书中所具体阐述及说明以外的其他方式来实施,且此为本领域技术人员所容易理解。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法