一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与流程
本发明属于有机功能材料制备技术领域,涉及一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法。
背景技术:
近年来,二氰基甲烯基苯并四氢呋喃衍生物因其优异的光电性能、红色荧光发射和良好的光稳定性等优点而受广大研究者的青睐。这类有机化合物常用于荧光分子探针,分子逻辑门,光增感剂和有机发光二极管等领域。但是由于存在分子间聚集淬灭现象,大多数二氰基甲烯基苯并四氢呋喃衍生物荧光量子产率低,因此大大限制了这类材料的广泛应用。目前报道提高二氰基甲烯基苯并四氢呋喃衍生物量子率的方法主要分为“引入枝状基团”和“引入更强的供电子和/或拉电子基团”。在二氰基甲烯基苯并四氢呋喃衍生物骨架上引入位阻较大的基团可以降低分子间π-π*堆积从而增强荧光量子产率。但是由于枝状基团的空间位阻效应和取代基效应,使得这类物质的再次修饰变得更加困难,因此限制了二氰基甲烯基苯并四氢呋喃衍生物合成与更深的应用。引入更强的供和/或拉电子基团可以增强二氰基甲烯基苯并四氢呋喃衍生物中的推拉效应,从而增强荧光量子产率。但是,利用这种方法制备的化合物最大荧光发射蓝移,位于红光区域(λem≤650nm)之外。此外,引入的供和/或拉电子基团占住了这些化合物的活性位点,使得引入其他官能团变得更加困难。
针对以上问题,因此发展一种简单、有效的方法降低分子间聚集淬灭效应提高二氰基甲烯基苯并四氢呋喃衍生物的荧光量子产率具有非常重要的意义。
技术实现要素:
针对目前制备高荧光量子产率二氰基甲烯基苯并四氢呋喃衍生物的方法存在荧光量子产率低、合成复杂、稳定性差、再次修饰困难等问题,本发明提供了一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法,这种方法以经典的包含羟基的二氰基甲烯基苯并四氢呋喃衍生物(简称DCM-OH)为原料,在羟基邻位引入一个氢键受体醛基即可得到一个具有分子内氢键作用的二氰基甲烯基苯并四氢呋喃衍生物(简称DCM-OH-CHO)。与传统的DCM-OH相比,该方法制备的DCM-OH-CHO具有更高的量子产率,较强的荧光稳定性,更适合再次修饰等特点。
为实现上述目的,本发明的技术方案是:
一种二氰基甲烯基苯并四氢呋喃衍生物,分子式为:
(简称DCM-OH-CHO)。
所述二氰基甲烯基苯并四氢呋喃衍生物的制备方法,包括以下步骤:
(1)以DCM-OH为原料,加入六次甲基四胺和酸,混合均匀,得混合液;所述混合液配方比例为:
DCM-OH 270毫克(0.8毫摩尔)
六次甲基四胺 56-1120毫克(0.4-8毫摩尔)
酸的体积 8-20毫升;
其中所述DCM-OH的分子式为:
所述酸选自盐酸、醋酸和三氟乙酸中的一种或几种;
(2)将上述混合液回流1-10小时;
(3)反应结束后,冷却至室温;中和残余酸液,萃取有机物质,干燥有机相,减压蒸馏除去溶剂,得到粗产物;
(4)所述粗产物用硅胶色谱分离提纯后得目标产物。
优选方案,步骤(1)所述混合液配方比例为:
DCM-OH 270毫克(0.8毫摩尔)
六次甲基四胺(HMT) 224-896毫克(1.6-6.4毫摩尔)
酸的体积 8-20毫升。
进一步优选,步骤(1)所述混合液配方比例为:
DCM-OH 270毫克(0.8毫摩尔)
六次甲基四胺(HMT) 224-560毫克(1.6-4.0毫摩尔)
酸的体积 8-20毫升。
优选方案,步骤(1)所述酸为三氟乙酸。
优选方案,步骤(2)是在回流下反应5小时至10小时。
优选方案,步骤(3)是采用饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机化合物。
优选方案,步骤(4)分离提纯是采用二氯甲烷作为洗脱剂。
所述粗产物为含一个和/或二个醛基的二氰基甲烯基苯并四氢呋喃衍生物;所述“和/或”的意思是含一个和二个醛基的二氰基甲烯基苯并四氢呋喃衍生物中的一种或两种。
在本发明中,二氰基甲烯基苯并四氢呋喃衍生物(DCM-OH)为修饰改性对象,所以反应原料中必须有DCM-OH。所使用的DCM-OH是按照文献报道方法合成,其结构经核磁共振表征确认。
本发明的制备方法受原料配料比,反应时间以及酸用量影响大。原料配料比过大,反应时间过长都会产生含二个醛基的副产物,使产物分离困难;原料配料比过低,反应时间过短导致DCM-OH-CHO的产率降低。酸的种类和用量也很关键,合适的酸可以缩短反应时间,提高反应产率;合适的用量可以避免原料浪费。
在本发明中,六亚甲基四胺用量和反应时间是关键,通过控制六亚甲基四胺用量和反应时间即可实现对DCM-OH-CHO的高效合成。当六亚甲基四胺用量为56毫克至224毫克时,DCM-OH-CHO的产率相对较低;当六亚甲基四胺用量控制在224毫克至896毫克之间时,产物DCM-OH-CHO相对较高,最优选在224毫克至560毫克之间产物收率高;当六亚甲基四胺用量超过896毫克后,DCM-OH-CHO被醛基化生成含二个醛基的二氰基甲烯基苯并四氢呋喃衍生物,产率降低。当反应时间控制在1小时至5小时内,有DCM-OH-CHO生成,但是还有部分原料未反应完;当时间在5小时至10小时内,DCM-OH已反应完全;反应时间超过10小时后,部分DCM-OH-CHO被醛基化生成含二个醛基的二氰基甲烯基苯并四氢呋喃衍生物,产率降低。
与现有技术相比,本发明的创新之处在于:
1、本发明成功合成了一种新的二氰基甲烯基苯并四氢呋喃衍生物,具有较强的荧光稳定性,更适合再次修饰。
2、本发明的方法相比传统引入空间位阻较大的基团制备二氰基甲烯基苯并四氢呋喃衍生物的方法,本方法可以得到高产率二氰基甲烯基苯并四氢呋喃衍生物DCM-OH-CHO。更重要的是,本方法制备的DCM-OH-CHO包含一个活性羟基和醛基,可以通过官能团间的转化合成其他二氰基甲烯基苯并四氢呋喃衍生物,增加了这种材料在其他领域中的实用性。
3、本发明的方法量子产率可以达到80%以上,且产物稳定性高:传统的二氰基甲烯基苯并四氢呋喃衍生物大多遭受分子内聚集淬灭效应,荧光量子产率不高。此外,已有文献对这些物质的荧光稳定性等参数没有进行深入研究。本发明中,在传统的二氰基甲烯基苯并四氢呋喃衍生物DCM-OH骨架中引入分子内氢键单元不仅降低分子间聚集淬灭效应,还提供一个稳定的刚性结构。因此,大大提高了其荧光量子产率和光稳定性。相比传统的DCM-OH,新合成的DCM-OH-CHO在不同水相和有机相比、pH值、有机溶剂、缓冲体系中显示了优异的荧光稳定性,因此在实际应用中将具有明显的优势。
附图说明
图1是本发明由DCM-OH为原料制备DCM-OH-CHO的合成路径图,由图1可知,经过简单一步合成可以制备得到DCM-OH-CHO;该合成在温和条件下进行,反应5个小时,即可得到目标产物(实施例1-3,产率为大于80%)。
图2是本发明所制备DCM-OH-CHO的氢谱,由图2可知,制备得到的产物比较纯净;低场11.4ppm处为羟基的质子共振信号,说明DCM-OH-CHO分子中存在羟基和醛基间的氢键作用。
图3是本发明所制备DCM-OH-CHO的红外光谱图,由图3可知,2820和2918cm-1处为羟基的特征峰,1630cm-1处为醛基的特征峰,说明本发明实施例成功制备得到DCM-OH-CHO。
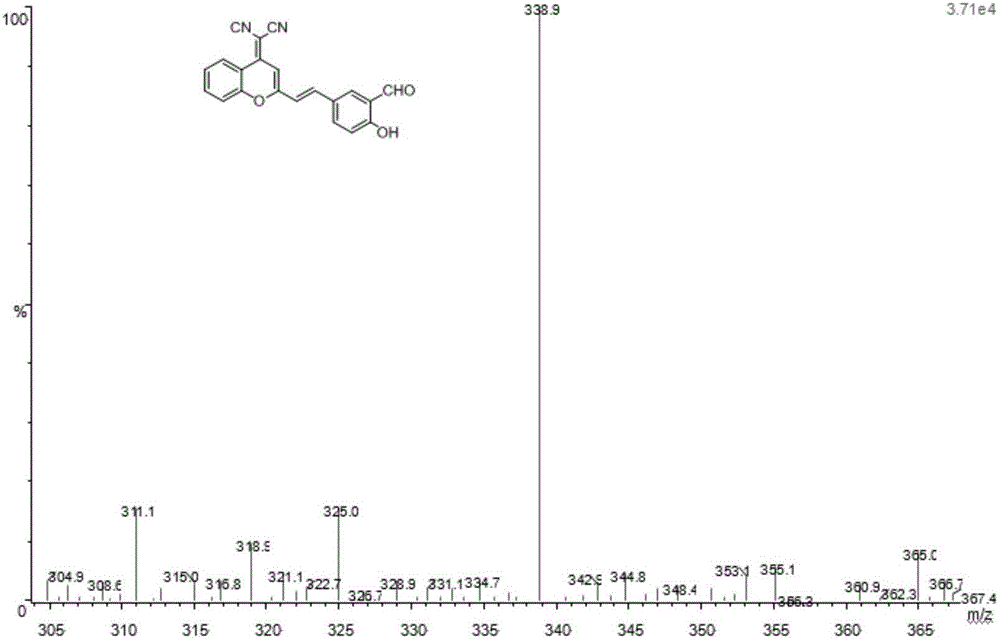
图4是本发明所制备DCM-OH-CHO的质谱,由图4可知,338.9处的峰为DCM-OH-CHO分子离子峰,说明本发明实施例成功制备得到DCM-OH-CHO。
图5是本发明所制备DCM-OH-CHO和传统DCM-OH的紫外-可见吸收光谱图,图5表明本发明所制备的DCM-OH-CHO摩尔吸光度高于传统DCM-OH。
图6是本发明所制备DCM-OH-CHO和传统DCM-OH的荧光发射光谱图,图6表明本发明所制备的DCM-OH-CHO荧光量子产率明显高于传统DCM-OH。
图7是本发明所制备DCM-OH-CHO和传统DCM-OH在不同水相与有机相比中的荧光发射光谱图,图7表明在传统DCM-OH分子中引入氢键受体醛基可以有效降低分子间聚集淬灭效应,提高其荧光量子产率。
图8是传统DCM-OH在不同pH中的荧光发射光谱图,图9是本发明所制备DCM-OH-CHO在不同pH中的荧光发射光谱图,图8和图9表明本发明所制备的DCM-OH-CHO抗pH性高于传统DCM-OH。
图10是本发明所制备DCM-OH-CHO和传统DCM-OH在不同有机溶剂中的荧光发射光谱图,图11是传统DCM-OH在不同有机溶剂中的荧光发射光谱图,图10和图11表明本发明所制备的DCM-OH-CHO在不同溶剂中的荧光稳定性高于传统DCM-OH。
图12是传统DCM-OH在不同缓冲体系中的荧光发射光谱图,图13是本发明所制备DCM-OH-CHO在不同缓冲体系中的荧光发射光谱图,图12和13表明本发明所制备的DCM-OH-CHO在不同缓冲体系中的荧光稳定性高于传统DCM-OH。
图14是本发明所制备DCM-OH-CHO和传统DCM-OH在不同光照射时长下的荧光发射光谱图,图14表明本发明所制备的DCM-OH-CHO继承传统DCM-OH优秀的抗光漂白性。
具体实施方式
下面结合实施例对本发明做进一步的说明。
实施例1
准确称取270毫克DCM-OH和224毫克六次甲基四胺于干燥的圆底烧瓶,然后加入8毫升三氟乙酸,加热回流5个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,剩余物用硅胶色谱柱分离提纯(二氯甲烷为洗脱剂)得到大量DCM-OH-CHO(产率高于84%)。产物的结构鉴定和性能鉴定请见附图1-14及附图说明。
实施例2
准确称取270毫克DCM-OH和224毫克六次甲基四胺于干燥的圆底烧瓶,然后加入15毫升三氟乙酸,加热回流5个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,剩余物用硅胶色谱柱分离提纯(二氯甲烷为洗脱剂)得到大量DCM-OH-CHO(产率为85%)。
实施例3
准确称取270毫克DCM-OH和224毫克六次甲基四胺于干燥的圆底烧瓶,然后加入20毫升三氟乙酸,加热回流5个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,剩余物用硅胶色谱柱分离提纯(二氯甲烷为洗脱剂)得到大量DCM-OH-CHO(产率为86%)。
实施例4
准确称取270毫克DCM-OH和56毫克六次甲基四胺于干燥的圆底烧瓶,然后加入8毫升三氟乙酸,加热回流5个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,剩余物用硅胶色谱柱分离提纯(二氯甲烷为洗脱剂)得到少量DCM-OH-CHO(产率为22%)。
实施例5
准确称取270毫克DCM-OH和896毫克六次甲基四胺于干燥的圆底烧瓶,然后加入8毫升三氟乙酸,加热回流5个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,粗产物进行核磁表征,证明生成部分DCM-OH-CHO(产率为41%)和部分含二个醛基的二氰基甲烯基苯并四氢呋喃衍生物。
实施例6
准确称取270毫克DCM-OH和224毫克六次甲基四胺于干燥的圆底烧瓶,然后加入8毫升三氟乙酸,加热回流1个小时;反应结束后,冷却至室温,TLC点板监控,发现大量原料未反应完全,生成DCM-OH-CHO的量少。
实施例7
准确称取270毫克DCM-OH和224毫克六次甲基四胺于干燥的圆底烧瓶,然后加入8毫升三氟乙酸,加热回流12个小时;反应结束后,冷却至室温,加入饱和碳酸氢钠溶液中和多余酸液,用二氯甲烷萃取有机物并用无水硫酸钠干燥;减压蒸馏除去溶剂,粗产物进行核磁表征,证明生成部分DCM-OH-CHO(产率为38%)和部分含二个醛基的二氰基甲烯基苯并四氢呋喃衍生物。
实施例8
用盐酸替代三氟乙酸,其他实验参数同实施例2。反应结束后,得到少量目标产物DCM-OH-CHO(产率为33%)。
实施例9
用醋酸替代三氟乙酸,其他实验参数同实施例2。反应结束后,得到少量目标产物DCM-OH-CHO(产率为37%)。
实施例10
用醋酸和盐酸混合液(体积比为1:2)替代三氟乙酸,其他实验参数同实施例2。反应结束后,得到少量目标产物DCM-OH-CHO(产率为35%)。
实施例11
用醋酸和盐酸混合液(体积比为2:1)替代三氟乙酸,其他实验参数同实施例2。反应结束后,得到少量目标产物DCM-OH-CHO(产率为37%)。
实施例12
用醋酸和盐酸混合液(体积比为1:1)替代三氟乙酸,其他实验参数同实施例2。反应结束后,得到少量目标产物DCM-OH-CHO(产率为39%)。
- 还没有人留言评论。精彩留言会获得点赞!
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 具有含硫取代基的杀有害生物活性杂环衍生物的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 靛红杂合喹唑啉类化合物合成的靛红衍生物及其在制备抗肿瘤药物中的应用的制造方法与工艺
- 一种杂环衍生物及使用该杂环衍生物的有机发光器件的制造方法与工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 一种含氮杂环衍生物及使用该含氮杂环衍生物的有机发光器件的制造方法与工艺
- 苯并二氮杂*衍生物的2-萘磺酸盐及晶型和它们的制备方法与制造工艺
- 一种二氰基甲烯基苯并四氢呋喃衍生物及其制备方法与制造工艺
- 苯并吲唑类衍生物及其制备方法、应用与制造工艺
- 一种二苯并环庚烷衍生物的制备方法与制造工艺
- Atropurpuran的苯并咪唑基和吗啉基衍生物组合物用于防治胰腺纤维化的制造方法与工艺
- 一种苯并咪唑衍生物镉配合物及其制备方法和应用
- 一种咪唑并吡啶巯乙酸类衍生物及其制备方法与应用
- Artalbic acid的O?(苯并咪唑基)乙基与O?(二羟乙胺基)乙基衍生物的组合物在抗病毒 ...的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备抗骨质疏松药物的制作方法
- 阿掏比克酸苯并咪唑基与二羟乙胺基衍生物的组合物用于制备防治胰腺纤维化药物的制作方法
- 一种具有抗氧化活性的芳基苯并呋喃类化合物及其制备方法和应用与流程
- 一种含苯并双环酮与乙氧磺隆的除草组合物及其应用的制造方法与工艺
- 一种6‑氯呋喃[3,2‑B]吡啶的合成方法与流程
- 一类苯并吡喃类化合物及其应用的制造方法与工艺
- 一种以二苯并环庚烯为核心的化合物及其在有机电致发光器件上的应用的制造方法与工艺
- 一种含苯并双环酮与乙草胺的除草组合物及其应用的制造方法与工艺
- 一种含苯并双环酮与噁草酮的除草组合物及其应用的制造方法与工艺
- 分解二噁英前体的催化剂筛选系统及其使用方法与流程
- 一种无卤热固性树脂组合物以及含有它的预浸料、层压板和印制电路板的制造方法与工艺
- 一种以二联二苯并五元杂环为骨架的有机化合物及其在OLED上的应用的制造方法与工艺
- 6,7,8,9‑四氢‑5H‑苯并[7]轮烯‑2‑烷基胺类化合物及其用途的制造方法与工艺
- 用于PHOLED的具有氮杂‑二苯并噻吩或氮杂‑二苯并呋喃核的新材料的制造方法与工艺
- 多孔ZSM‑5沸石与γ‑Al2O3复合材料的合成及制备加氢脱硫催化剂的制造方法与工艺
- 一种含有氟噻虫砜和五氟虫腙的杀虫组合物的制造方法与工艺
- 苯并双(噻二唑)衍生物、包含其的油墨、以及使用了其的有机电子装置的制造方法
- 苯并噻二唑胺的制造方法与工艺
- 用于有机电致发光器件的材料的制造方法与工艺
- 用于有机电致发光器件的材料的制造方法与工艺
- 一种苯并噻咯类衍生物及其制备方法和有机发光器件与制造工艺
- 苯并噻吩并咔唑类衍生物及其制备方法和有机发光器件与制造工艺