使用基于水凝胶的液滴进行单细胞基因组测序的制作方法
交叉引用本申请要求2016年12月21日提交的美国临时专利申请第62/437,605号的权益,其以全文引用的方式并入本文中。政府支持本发明是在政府支持下在由美国国家卫生研究院(nationalinstitutesofhealth)授予的授权号ar068129、r01eb019453和r21hg007233下;由美国国家科学基金会(nationalsciencefoundation)授予的授权号1253293下;由国防部高级研究计划局(departmentofdefense,defenseadvancedresearchprojectsagency)授予的授权号hr0011-12-c-0065下;以及由空间和海战系统中心(spaceandnavalwarfaresystemscenter)授予的授权号n66001-12-c-4211下制成的。政府拥有本发明的某些权利。
背景技术:
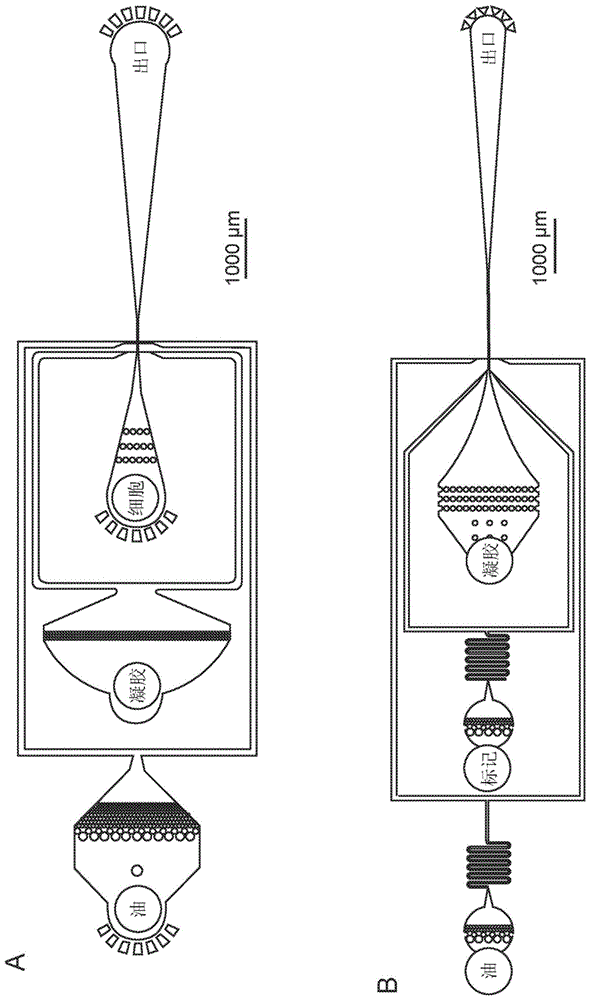
:将单细胞测序施加于异质系统时常见的挑战是其通常含有大量细胞:厘米大小的肿瘤可能含有亿万个突变的癌细胞,而一毫升的海水可能含有数百万个微生物。此外,每个细胞都具有少量的dna,因此难以准确地扩增和测序如此多单细胞。基于光学镊子、流式细胞术、微流体、凝胶包封和虚拟微流体的方法可以分离和处理数百个单细胞用于测序,但这构成大多数群落的一小部分。取样的稀疏性限制了可以解决的问题,大多数研究结果与最丰富的亚种群有关。例如,常见的环境群落含有>1500个分类群,稀有分类群以<0.1%存在,其中大多数是单细胞测序错过的;事实上,捕获这些细胞的难度是“微生物暗物质”的基础-人们认为存在大量物种,但这种物质从未被表征过。因此,一种可以显著增加通过单细胞测序而测序的细胞数量的方法将影响生物学中广泛的问题,其中异质性是重要的。技术实现要素:本公开提供了超高通量的单细胞基因组测序方法,在本文中称为“sic-seq”,所述方法包括在熔融凝胶液滴中包封单细胞以促进微凝胶中的大量细胞裂解和基因组dna的纯化。还提供了用于实践本发明方法的系统和装置。附图说明当结合附图阅读时,从以下详细描述中可最佳地理解本发明。附图中包括以下图:图1提供了根据本公开的一个实施例的微流体装置的示意图,其用于a)产生条形码液滴并将细胞包封在微凝胶中;b)用标记试剂重新包封凝胶;和c)将凝胶液滴与条形码液滴和pcr液滴合并。图2提供了根据本公开的一个实施例的实例sic-seq工作流程的示意图。将单细胞包封在微凝胶中,并将基因组纯化并片段化,例如,在一系列清洁剂和酶洗涤中。然后用核酸标记基因组片段,所述核酸例如dna,每个液滴独有的条形码序列。合并得到的条形码基因组片段并测序,产生可以基于共享条形码由单细胞分组的读段。读段组包含单细胞的低覆盖度基因组的数据库,其可以例如使用计算机模拟血细胞计数进行分析。图3a-3c提供了根据本公开的一个实施例的以产生sic-seq库的微流体和生物化学工作流程的示意图。图3a)通过使用流动聚焦液滴制造器用pcr试剂包封有限稀释下的随机dna寡核苷酸来产生条形码液滴。对液滴进行热循环,产生含有每约9个空液滴的独特条形码序列的克隆群体的液滴(sybr染色用于可视化)。图3b)用熔融凝胶(例如熔融琼脂糖)在有限稀释下包封细胞,例如细菌,以产生含有微凝胶的单细胞,例如含有琼脂糖微凝胶的单细胞。通过一系列大量洗涤(例如清洁剂和酶洗涤)纯化单细胞基因组。然后将纯化的单细胞基因组重新包封并标记,例如标记(tagmented)。图3c)将标记的,例如标记的,含基因组的微凝胶与含有条形码和核酸扩增试剂(例如pcr试剂)的液滴合并。在热循环期间,条形码剪接到标记的,例如标记的基因组片段上,产生嵌合分子,所述嵌合分子由附接到易于大规模平行测序的细胞基因组的随机片段的条形码组成。图4描绘了显微镜图像和表征琼脂糖微凝胶内基因组片段扩散的图。a)sybr染色用于监测标记之前和之后微凝胶中的基因组的扩散。b)在室温下两天后,通过离心沉淀微凝胶,并从微凝胶和上清液中萃取dna,并使用qubitdsdna高灵敏度测定和生物分析仪高灵敏度dna芯片定量。由于所用的转座酶与基因组的化学计量比相对较低,片段大小的变化相对较小。c)使包封的基因组与转座酶与基因组的更高的化学计量比反应,并在生物分析仪高灵敏度芯片上显现,以显示凝胶包封的基因组的片段化效率。图5a-5e描绘了显示在人工构建的微生物群落上sic-seq的性能的图。图5a)每个条形码组中的读段分布。图5b)每个条形码组的纯度的直方图,其定义为映射到所述组的最映射物种的读段分数。图5c)对于每个物种使用从左到右计算每个物种的相对丰度估计:使用bowtie2(bowtie2读段)分类的读段、使用bowtie2(bowtie2条形码)分类的条形码,和使用kraken(kraken读段)分类的读段。图5d)对于每种微生物从所有条形码组聚集的读段的相对覆盖度分布。图5e)由相对覆盖度分组的覆盖度直方图。参见用于其它物种的覆盖图6a和6b。图6a和6b描绘了从已知微生物的混合群落测序获得的sic-seq读段的分布。图6a)用于10kb箱的sic-seq验证数据集中的枯草芽孢杆菌(baccilussubtilis)和酿酒酵母(saccharomycescerevisiae)参考基因组的聚集覆盖度。图6b)映射具有>2000个读段的随机选择的条形码葡萄球菌(staphylococcus)组的读段的位置。图7描绘了说明sic-读段数据库的实例框架的示意图。含有如序列、读段id和分类特性的读段存储在条形码组中,所述条形码组含有如纯度和分类的特性。所示的序列仅用于举例而非限制。图8说明了用于显示如本文实例中所述的计算机模拟血细胞计数的海洋微生物群落。a)按条形码组的sic-读段数据库的分类丰度。b)在属水平下的数据库中条形码组的纯度的分布。图9a-9c示出了对回收自旧金山海岸线的海洋群落的sic-seq的应用。图9a)根据宿主微生物属的抗生素抗性(ar)基因的分布。使用计算机模拟血细胞计数,推导出ar基因与分类学分类的关联。连接线的不透明度反映了数据库中检测到的交互次数。图9b)在群落中检测到的每个属中的毒力因子的相对丰度。毒力比是由毒力因子观察到的条形码组数与所述物种的总条形码组数之间的比率,归一化为0至1的标度。图9c)作为热图绘制的群落中细菌分类群之间转导的相对潜力。图10描绘了通过模拟分离的菌株的基因组序列的读段而获得的参考数据,用于与本文实例中描述的海洋微生物群落中的数据进行比较。a)公共数据库中全基因组测序菌株的抗生素抗性网络。b)针对公开可用菌株计算的毒力因子比。图11描绘了显示每个条形码组的基因组覆盖度的平均值和分布的图,其绘制为每个物种的洛伦兹曲线。图12描绘了显示10细胞对照实验中条形码组的基因组大小-归一化纯度得分的图。使用相同方法使用针对每个相应物种测序的基因组分数而不是原始读段来计算基因组大小-归一化纯度得分。图13描绘了显示针对每个物种分别绘制的条形码组的纯度得分的图。图14描绘了显示a)纯度<80%的条形码组,b)纯度>80%的条形码组中的下一个最丰富物种的纯度得分的图。在<80%纯度的条形码组中,下一个最丰富的物种的纯度得分往往高达约20%至50%,这反映出这两个物种表示条形码组中的大多数读段,这表明这些条形码组表示双重包封。具有100%纯度的条形码组未在图中表示。图15描绘了显示葡萄球菌、芽孢杆菌和酵母菌属组成的人工构建的微生物群落上的sic-seq性能的图。对于每个物种使用从左到右计算每个物种的相对丰度估计:没有条形码的标记基因计数(metaphlan)、条形码计数(条形码)、细胞包封后显微镜下的人工计数(显微镜计数),以及培养时(理论值)。图16描绘了显示合成微生物群落中物种的所有条形码组的聚集基因组覆盖度的图。低丰度下的物种显示频繁的退出,其特征是图中的凹陷,但很少观察到以尖峰为特征的系统偏差的情况。图17提供了使用非对称数字液滴pcr条形码稠合至使用单循环延伸的malbac扩增子的条形码基因组dna的工作流程的示意图。图18提供了使用对称数字液滴pcr条形码稠合至使用重叠延伸随后多轮pcr的malbac扩增子的条形码基因组dna的工作流程的示意图。图19提供了描绘使用条形码malbac引物产生组合条形码环状扩增子的分子条形码方案的示意图。图20提供了描绘用于产生组合条形码基因组dna扩增子的微流体工作流程的示意图。图21提供了用于产生含有两种水相的混合物的30μm油包水乳液的微流体装置的示意图。对于数字条形码液滴的产生,装置在插入入口1时操作。图22提供了用于将malbac扩增的细胞液滴与数字pcr条形码液滴合并的微流体装置的示意图。阴影矩形表示合并区域,其中经受高电场梯度的液滴合并。图23提供了由示例性malbac条形码融合反应产生的dna产物的代表性电泳图(在大小选择之前)。图24提供了由示例性malbac条形码融合反应产生的dna产物的代表性电泳图(在大小选择之后)。图25描绘了显示实例2中所有条形码组的条形码组纯度的频率的直方图。插图以对数标度绘制。实验中所有条形码组的平均纯度为0.950(最小组大小为50个读段)。图26描绘了对于实例2的条形码组纯度对条形码组中的读段的散布图。每个点表示一个条形码组。仅显示具有最少500个读段的条形码组。图27描绘了表示条形码组的基因组全覆盖图(物种:枯草芽孢杆菌)。沿枯草芽孢杆菌基因组的所有位置放置一个点,其中来自条形码组的读段对齐。具体实施方式本公开提供了对单细胞基因组dna进行测序以分析例如宏基因组、拷贝数变体和复合物生物样品的基因曲线的方法。本文描述促进基因组dna的单细胞和后续测序的群体的高通量处理的方法还提供了用于实践本发明方法的系统和装置。在更详细描述本发明之前,应当理解,本发明不限于描述的具体实施方式,因为这种可以改变。还应了解,本文中所使用的术语仅出于描述特定实施例的目的,且不希望具有限制性,原因是本发明的范围仅由所附权利要求书限定。在提供数值范围的情况下,应理解还专门公开所述范围的上限与下限之间的每个中间值,直到下限单位的十分之一(除非上下文另外明确规定)。所陈述的范围内的任何所陈述值或中间值与那个所述范围内的任何其它所陈述值或中间值之间的每个更小范围涵盖于本发明中。这些较小范围的上限和下限可以独立地包括在所述范围内或排除在其外,并且每个范围(其中两个极限之一、没有一个、或都包括在所述较小的范围内)也涵盖在本发明内,服从所述范围中任何特别排除的限值。在所陈述范围包括限值中的一个或两个的情况下,排除所包括的那些限值中的任一个或两个的范围也包括于本发明中。除非另外定义,否则本文所使用的所有技术和科学术语均具有与本发明所属领域的一般技术人员通常所理解相同的含义。尽管与本文所述的那些类似或等效的任何方法和材料也可用于实践或测试本发明,但是一些潜在的和示例性的方法和材料现在可以被描述。本文提及的任何和所有公开案均以引用的方式并入本文中,从而公开和描述与所引用公开案相关的方法和/或材料。应当理解,在有矛盾的情况,本公开取代并入的出版物的任何公开内容。应注意,除非上下文另外明确规定,否则如本文和所附权利要求书中所用的单数形式“一(a/an)”以及“所述”包括多个指示物。因此,例如,除非上下文另外明确规定,否则提及的“液滴”包括多个此类液滴。还应注意,可以撰写权利要求以排除任何要素,例如任何任选的要素。因此,此陈述旨在作为使用与权利要求要素的叙述相关的“单独”,“仅”等排他性术语或使用“否定”限制的先行基础。本文中论述的出版物仅仅提供其在本申请的申请日之前的公开内容。另外,所提供的公开日期可以不同于可能需要独立确认的实际公开日期。对于本文中任何术语的公开或定义或使用与以引用的方式并入本文中的应用或出版物中的任何术语的公开或定义或使用相冲突的程度下,则本申请应受控制。如本领域的技术人员在阅读本发明之后将显而易见,本文中所描述和说明的每个个别实施例具有离散的组件和特征,所述特征可容易与其它若干实施例中的任一个的特征分离或与其组合而不脱离本发明的范围或精神。任何所述方法均可以所述事件的顺序或以逻辑上可能的任何其它顺序来进行。本文所用的术语“核酸条形码序列”,“核酸条形码”,“条形码”等是指具有可用于识别和/或区分其中核酸条形码从一个或多个第二分子缀合的一个或多个第一分子的序列的核酸。核酸条形码序列通常较短,例如,长度为约5至20个碱基,并且可以与所关注的一个或多个靶标分子或其扩增产物缀合。核酸条形码序列可以是单链或双链的。术语“核酸”、“核酸分子”、“寡核苷酸”和“聚核苷酸”可互换使用,并且是指任何长度的核苷酸的聚合形式,即脱氧核糖核苷酸或核糖核苷酸,或其类似物。术语涵盖例如dna,rna及其改性形式。聚核苷酸可以具有任何三维结构,并且可进行任何已知或未知的功能。聚核苷酸的非限制性实例包括基因、基因片段、外显子、内含子、信使rna(mrna)、转移rna、核糖体rna、核酶、cdna、重组聚核苷酸、支链聚核苷酸、质粒、载体、任何序列的分离的dna、控制区,任何序列的分离的rna、核酸探针和引物。核酸分子可以是线性的或环状的。术语“核酸序列”或“寡核苷酸序列”是指连续的核苷酸碱基串,并且特别地,上下文还指核苷酸碱基在寡核苷酸中出现时彼此相关的特定位置。类似地,术语“多肽序列”或“氨基酸序列”是指连续的氨基酸串,并且特别地,上下文还指氨基酸在多肽中出现时彼此相关的特定位置。如本文所用,术语“分离的”,当在分离的细胞的上下文中使用时,是指所关注的细胞,其处于与细胞天然存在的环境不同的环境中。“分离的”意指包括在样品内的细胞,这些样品基本上富含所关注的细胞和/或其中所关注的细胞为部分或基本上纯化的。本文使用的术语“液滴(droplets/droplet)”等是指能够包封和/或含有如本文所述的一种或多种单细胞和/或如本文所述的一种或多种条形码的基于乳液的隔室。液滴可包括第一流体相,例如水相(例如水或水凝胶),其以与第一流体相不混溶的第二流体相(例如油)为界。在一些实施例中,第二流体相是不混溶的相载体流体。因此,根据本公开的液滴可以以油包水乳液的形式提供。此外,本文所使用的术语“液滴”在“固化微凝胶液滴”的上下文中是指含有液滴(droplet/droplets)的熔融凝胶,其中熔融凝胶已经固化,留下由不混溶的相膜(例如油膜)包围的固化微凝胶。如结合本发明方法、装置和/或系统所使用或产生的液滴可以是球形的,或者其可以具有任何其它合适的形状,例如卵形或椭圆形。如本文所述的液滴可包括液相和/或固相材料。在一些实施例中,根据本公开的液滴包括凝胶材料。在一些实施例中,本发明液滴具有为或约1.0μm至1000μm(包括端值)的尺寸(例如,直径),如1.0μm至750μm、1.0μm至500μm、1.0μm至100μm、1.0μm至10μm或1.0μm至5μm(包括端值)。在一些实施例中,如本文所述的液滴具有为或约1.0μm至5μm、5μm至10μm、10μm至100μm、100μm至500μm、500μm至750μm或750μm至1000μm(包括端值)的尺寸(例如,直径)。此外,在一些实施例中,如本文所述的液滴具有在约1fl至1nl(包括端值)范围内的体积,如1fl至100pl、1fl至10pl、1fl至1pl、1fl至100fl或1fl至10fl(包括端值)。在一些实施例中,如本文所述的液滴具有1fl至10fl、10fl至100fl、100fl至1pl、1pl至10pl、10pl至100pl或100pl至1nl(包括端值)的体积。另外,如本文所述的液滴可具有大小和/或形状,使得其可在微流体装置中、在微流体装置上或由微流体装置产生和/或由微流体装置流动或由微流体装置施加。如本文所用,术语“载体流体”是指配置或选择为含有一个或多个液滴的流体,如本文所述。载体流体可以包括一种或多种物质,并且可以具有一种或多种特性,例如粘度,其允许载体流体流过微流体装置或其一部分。在一些实施例中,载体流体包括例如:油或水,并且可以是液相或气相。如权利要求中所使用的,术语“包含”与“包括”、“含有”和“特征在于”同义,是包括性的或开放式的,并且不排除另外的、未叙述的要素和/或方法步骤。“包含”是一个专门术语,意指存在所指定的要素和/或步骤,但是可以添加其它要素和/或步骤,并且仍然在相关主题的范围内。如本文所用,短语“由……组成”排除未具体叙述的任何要素、步骤和/或成分。例如,当短语“由……组成”出现在权利要求主体的一个条款中,而不是紧跟在前言之后时,其仅限制所述条款中阐述的要素;其它要素不排除在整个权利要求之外。如本文所用,短语“基本上由……组成”将相关公开或权利要求的范围限制于指定的材料和/或步骤,以及不会实质上影响所公开的和/或所要求的主题的基本和新颖特征。就术语“包含”、“基本上由……组成”及“由……组成”而言,在本文中使用这三个术语之一的情况下,所公开的主题可以包括使用另外两个术语中的任一个。方法如上概述,本公开提供超高通量的单细胞基因组测序方法,在本文中称为“sic-seq”,所述方法包括在熔融凝胶液滴中包封单细胞以促进微凝胶中的大量细胞裂解和基因组dna的纯化。这些方法有助于对单细胞基因组dna进行测序以分析例如宏基因组、拷贝数变体和复合物生物样品的基因曲线。用于测序单细胞基因组dna的方法本公开提供了对单细胞基因组dna进行测序的方法。本公开的方法提供单细胞基因组的超高通量测序。在一些实施例中,液滴微流体用于分离、片段化和条形码化细胞群体的单细胞基因组,使得通过条形码对读段进行分组来回收单细胞基因组dna。单细胞的分离:使用本文所述的方法,可以以液滴分离单细胞。在一些实施例中,将单细胞包封在液滴中,这可以促进纯化基因组dna的过程,而不混合单个单细胞的基因组内容物。在一些实施例中,使用包含液滴发生器的微流体装置实现将单细胞包封在液滴中。例如,在足以实现通道中细胞的惯性排序的条件下,单细胞群体可以流过微流体装置的通道,微流体装置包括与通道流体连通的液滴发生器,从而提供周期性的将细胞注入液滴发生器中以将单细胞包封在单个液滴中。在一些实施例中,将单细胞包封在液滴中的方法包含添加不混溶的相流体,例如油,以产生各自含有单细胞的液滴乳液。使用微流体液滴发生器的细胞包封的另外描述可见于例如美国专利申请公开第20150232942号中,其公开以全文引用的方式并入本文中。在一些实施例中,其中包封单细胞的液滴包含聚合物材料。例如,合适的聚合物材料可包括互穿聚合物网络(ipn);合成水凝胶;半互穿聚合物网络(sipn);热响应性聚合物等。例如,在一些实施例中,合适的聚合物包含聚丙烯酰胺和聚(乙二醇)(peg)的共聚物。在一些实施例中,合适的聚合物包含聚丙烯酰胺和peg的共聚物,并且还包含丙烯酸。在一些实施例中,其中包封单细胞的液滴可以是微凝胶液滴。在此类实施例中,微凝胶液滴可以是包含水凝胶聚合物的水凝胶液滴。合适的水凝胶聚合物可包括但不限于以下:乳酸、乙醇酸、丙烯酸、1-甲基丙烯酸羟乙基酯(hema)、甲基丙烯酸乙酯(ema)、丙二醇甲基丙烯酸酯(pema)、丙烯酰胺(aam)、n-乙烯基吡咯烷酮、甲基丙烯酸甲酯(mma)、甲基丙烯酸缩水甘油酯(gdma)、甲基丙烯酸乙二醇酯(gma)、乙二醇、富马酸等。一些水凝胶聚合物需要使用交联剂。常用的交联剂包括四乙二醇二甲基丙烯酸酯(tegdma)和n,n'-亚甲基双丙烯酰胺。水凝胶液滴可以是均聚的,或者可以包含两种或更多种上述聚合物的共聚物。示例性水凝胶液滴包括但不限于聚(环氧乙烷)(peo)和聚(环氧丙烷)(ppo)的共聚物;pluronic.tm.f-127(标称式eo100-po65-eo100(其中eo是环氧乙烷且po是环氧丙烷)的peo和ppo的双官能嵌段共聚物);泊洛沙姆(poloxamer)407(由通过聚(乙二醇)的两个亲水性嵌段侧接的聚(丙二醇)的中心嵌段组成的三嵌段共聚物);聚(环氧乙烷)-聚(环氧丙烷)-聚(环氧乙烷)共聚物,其标称分子量为12,500道尔顿,且peo:ppo比为2:1);聚(n-异丙基丙烯酰胺)-碱基水凝胶(基于pnipaam的水凝胶);pnipaam-丙烯酸共聚物(pnipaam-共-aac);聚(甲基丙烯酸2-羟乙酯);聚(乙烯基吡咯烷酮)等。在本文描述的方法中特别有用的是能够从一种状态转变为另一种状态的微凝胶液滴,例如,从液态转变为固态。在一些实施例中,微凝胶液滴是包含水凝胶聚合物的水凝胶液滴,其中水凝胶聚合物是热响应性聚合物。热响应性聚合物通常表现出其物理特性随温度的变化。例如,热响应性聚合物可在一定温度下表现出体积相变,这导致溶剂化状态的突然变化。适用于本公开的方法的热响应性聚合物可包括在加热时变得可溶的那些。例如,琼脂糖,例如低凝胶温度琼脂糖,可适用于本文所述的方法。在一些实施例中,合适的热响应性聚合物,例如合适的琼脂糖,具有约20℃至约40℃的凝胶点,例如约25℃至约35℃,例如约30℃。热响应性聚合物可具有与其熔点不同的凝胶点。如本文所用,热响应性聚合物的术语“凝胶点”是指液体热响应性聚合物固化(例如转变成固态)的温度。如本文所用,热响应性聚合物的术语“熔点”或“熔融温度”是指固体热响应性聚合物熔融下(例如转变成液态)的温度。在一些实施例中,合适的热响应性聚合物,例如合适的琼脂糖,具有约5℃至约45℃,例如约5℃至约10℃、约10℃至约15℃、约15℃至约20℃、约25℃至约30℃、约30℃至约35℃、约35℃至约40℃、约40℃至约45℃,例如约20℃的凝胶点。在一些实施例中,合适的热响应性聚合物,例如合适的琼脂糖具有约20℃的凝胶点。在一些实施例中,合适的热响应性聚合物,例如合适的琼脂糖具有约60℃至约95℃,例如约60℃至约65℃、约65℃至约70℃、约70℃至约75℃、约75℃至约80℃、约80℃至约85℃、约85℃至约90℃、约90℃至约95℃,例如约60℃的熔点。在一些实施例中,合适的热响应性聚合物,例如合适的琼脂糖具有约60℃的熔点。在一些实施例中,单细胞可以包封在熔融凝胶液滴中,例如熔融琼脂糖凝胶液滴中,其可以固化成固化微凝胶液滴。在一些实施例中,熔融琼脂糖凝胶液滴通过冷却固化。在一些实施例中,微凝胶液滴可包含在交联时转化为固态的聚合物。例如,水凝胶液滴可包含丙烯酰胺并在化学和/或光交联时固化。例如,微凝胶液滴可包含聚(乙二醇)(peg)并在化学和/或光交联时固化。在一些实施例中,水凝胶液滴可包含藻酸盐,其在添加钙时固化。在一些实施例中,用于本文所述方法的微凝胶液滴包括或形成固化微凝胶,其具有大小设定成将核酸保留在固化微凝胶内的孔。在一些实施例中,固化微凝胶包括大小设定成将核酸保留在固化微凝胶内的孔,但允许其它材料移入和移出固化微凝胶。例如,大分子大分子(例如,基因组dna)保留在固化微凝胶中,而其它材料例如脂质和蛋白质能够移出固化微凝胶,例如,由于一个或多个洗涤步骤。固化微凝胶的孔径是微凝胶类型和使用的浓度的函数。在一些实施例中,固化微凝胶是固化琼脂糖微凝胶,其孔径是所用琼脂糖类型和浓度的函数。在一些实施例中,在约1.5%至约2%浓度下,例如在约1.3%至约1.5%下、在约1.4%至约1.6%下、在约1.5%至约1.7%下、在约1.6%至约1.8%下、在约1.7%至约1.9%下、在约1.8%至约2.0%下、在约1.9%至约2.1%下由微凝胶配制物制成的固化微凝胶的孔径可具有在约50nm至约150nm,例如,约30nm至约50nm、约50nm至约70nm、约70nm至约90nm、约90nm至约110nm、约110nm至约130nm、约130nm至约150nm、约150nm至约170nm范围内的孔径。本领域普通技术人员将能够测定微凝胶孔径。例如,微凝胶孔径可通过narayanan等人,《物理期刊:会议系列(journalofphysics:conferenceseries)》,2006,28:83-86中描述的方法测定,其公开以引用的方式并入本文中,因此,在一些实施例中,如由本公开提供的测序单细胞基因组dna的方法包括:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞。用于对来自本文提供的单细胞群体的单细胞基因组dna进行测序的方法可用于测序来自复杂细胞混合物的单细胞基因组dna。在一些实施例中,单细胞群体可以是同质的或异质的。在一些实施例中,单细胞群体可包括真核细胞(例如,哺乳动物细胞、真菌细胞等)或原核细胞(例如细菌细胞)或其组合。在一些实施例中,单细胞群体可以从多种来源获得,例如通过静脉穿刺收集的血液样品、通过手指穿刺收集的血液样品、通过腰椎穿刺收集的脑脊髓液样品、环境样品等。单细胞基因组dna的纯化:根据本文所述方法的实施例,单个单细胞的基因组dna被大量纯化,同时保持来自不同固化微凝胶中的不同细胞的基因组dna的分离。在一些实施例中,通过将单细胞包封在可以固化的微凝胶液滴中来促进单细胞基因组dna的大量纯化。例如,从单细胞群体中纯化单细胞基因组dna的方法包括将单细胞的群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞。然后固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴。方法包括破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体,并将大量固化的微凝胶群体暴露于裂解条件。在一些实施例中,将固化的微凝胶群体大量暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件。在一些实施例中,裂解条件包括使固化的微凝胶群体与裂解酶接触。本领域普通技术人员将认识到,可以使用不同的裂解酶,这取决于固化微凝胶中的细胞类型。例如,可以使用一种或多种裂解酶裂解细菌细胞,所述裂解酶包括例如无色肽酶、唇形酶、溶葡球菌酶、溶菌酶、变溶菌素等。可以使用一种或多种裂解酶裂解酵母细胞,所述裂解酶包括例如酵母裂解酶(zymolyase)、细胞裂解酶、glucanex、溶细胞酶(lyticase)等。可以使用一种或多种裂解酶裂解植物细胞,所述裂解酶包括例如纤维素、果胶酶(pectinase)、果胶酶(pectolyase)等。可以使用一种或多种裂解酶裂解哺乳动物细胞,所述裂解酶包括例如破伤风溶血素、α-溶血素、链球菌溶血素o等。本领域普通技术人员将能够从多种裂解酶中选择最适合裂解所关注的细胞的裂解酶的一种或组合。在一些实施例中,所公开的方法具体包括使大量固化的微凝胶群体与两种或更多种不同的裂解酶接触,例如,其中两种或更多种不同的裂解酶能够裂解不同的细胞类型。这种接触可以同时发生或在单独的时间步骤中发生,例如通过一个或多个洗涤步骤分开。在一些实施例中,细胞裂解包括使大量固化的微凝胶群体与一种或多种裂解酶(例如裂解酶的混合物)接触,并将固化微凝胶温育足以裂解细胞的一段时期,例如约5分钟至约24小时(包括端点),例如约10分钟至约24小时、约20分钟至约24小时、约30分钟约至约24小时、约40分钟至约24小时、约50分钟至约24小时、约1小时至约24小时、约3小时至约24小时、约6小时至约24小时、约9小时至约24小时、约12小时至约24小时、约15小时至约24小时、约18小时至约24小时,或约21小时至约24小时。在一些实施例中,细胞裂解包括使大量固化的微凝胶群体与一种或多种裂解酶(例如裂解酶的混合物)接触,并将固化微凝胶温育约10分钟至约20分钟、约20分钟至约30分钟、约30分钟至约1小时、约1小时至约3小时、约3小时至约6小时、约6小时至约9小时、约9小时至约12小时、约12小时至约15小时、约15小时至约18小时、约18小时至约21小时,或约21小时至约24小时。在一些实施例中,将固化的微凝胶群体与裂解酶的混合物一起温育过夜以裂解含于固化微凝胶内的细胞。在一些实施例中,从单细胞群体纯化单细胞基因组dna的方法包括使固化的微凝胶群体与一种或多种清洁剂接触以溶解含于固化的微凝胶群体内的细胞物质。例如,清洁剂可以溶解在细胞裂解时释放的膜脂质。适用于本公开方法的清洁剂包括本领域熟知的那些。常用清洁剂包括:十二烷基硫酸钠(sds)、sds月桂基、sdsc12、tritonx-100、tritonx-114、np-40、tween-20、tween-80、辛基葡糖苷、辛硫基葡糖苷、3-((3-胆酰胺基丙基)二甲氨基)-1-丙磺酸盐(chaps)、十二烷基硫酸锂等。在一些实施例中,所公开的方法具体包括使大量固化的微凝胶群体与两种或更多种不同的清洁剂接触。这种接触可以同时发生或在单独的时间步骤中发生,例如通过一个或多个洗涤步骤分开。在一些实施例中,从单细胞群体纯化单细胞基因组dna的方法包括使固化的微凝胶群体与蛋白酶接触以消化含于固化的微凝胶群体内的细胞蛋白质。适用于本公开的方法的蛋白酶包括本领域中众所周知的那些,例如丝氨酸蛋白酶、枯草杆菌蛋白酶型蛋白酶,例如蛋白酶k、brofasin等。在一些实施例中,在足以消化细胞蛋白质的条件下,例如在50℃下30分钟或足以消化含于固化的微凝胶群体内的细胞蛋白质的任何其它合适的温度和时间,将固化的微凝胶群体与蛋白酶k一起温育。如本文所述,在固化的微凝胶群体内细胞裂解后,大分子量大分子(例如,基因组dna)被捕获在固化微凝胶内。因此,来自一个细胞的基因组dna不与另一个细胞的基因组dna混合,有效地将每个单个的单细胞的基因组dna区分到其各自的固化微凝胶中。由于固化微凝胶的孔隙度,较小大小的分子如裂解酶、蛋白酶和清洁剂能够使固化微凝胶自由地进入,例如消化蛋白质、消化细胞壁的片段和溶解脂质。细胞裂解成功后,可以纯化基因组dna。在一些实施例中,洗涤固化的微凝胶群体以去除裂解酶和/或清洁剂和其它化学物种,其可抑制下游分子生物学反应,例如pcr反应。通过使固化的微凝胶群体与洗涤缓冲液接触来洗涤固化的微凝胶群体。在一些实施例中,洗涤固化微凝胶包括使固化的微凝胶群体与一系列洗涤缓冲液接触。在一些实施例中,洗涤缓冲液包括tween-20和乙醇,但是可以使用本领域已知的任何合适的洗涤缓冲液。因此,如由本公开提供的测序单细胞基因组dna的方法包括:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;和从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化的基因组dna的固化的微凝胶群体;本公开中提供的方法的一个重要优点是将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件引起应用可能与液滴中进行的裂解方法不相容的苛刻(严格)裂解条件。例如,使用强清洁剂(例如十二烷基硫酸钠)和化学品,其由本文提供的方法提供,可使油包水乳液液滴不稳定。因此,在本方法中裂解期间使用强清洁剂和化学品的能力可以允许研究更多样化的细胞类型。此外,将大量固体微凝胶的群体暴露于裂解条件引起不同裂解条件的高效应用,例如,在单独的步骤中(例如,通过一个或多个洗涤步骤分开),其被设计或足以裂解可存在于原始细胞群体中的不同的细胞类型。片段化和标记基因组dna所公开的方法可包括片段化基因组dna的步骤,例如,允许其用现有测序平台测序的长度,所述测序平台通常具有有限的阅读长度。可以以多种方式实现片段化,并且可以将其施加到扩增的或非扩增的核酸靶标。例如,可以将能够使dna片段化的酶如或其它核酸酶引入如本文所述的微凝胶液滴、固化微凝胶液滴和/或固化微凝胶,并且微凝胶液滴、固化微凝胶液滴和/或固化微凝胶经受足以片段化的条件。能够使dna片段化的合适的酶可包括例如dna酶i、微球菌核酸酶、dna酶iii和导致片段化dna的任何其它核酸酶,包括具有序列特异性催化的核酸酶。替代地,可以使用化学方法,如包括酸、活性氧物种等。降解dna的生物也可以通过将其包括在微凝胶液滴、固化微凝胶液滴和/或具有核酸的固化微凝胶中来使用。还可以使用物理方法,如在微凝胶液滴、固化微凝胶液滴和/或固化微凝胶中通过核酸流动产生的剪切。也可以使用其它方法在核酸上进行多种操作,包括片段化。例如,转座子可用于将序列插入或附接到核酸中,通常在过程中将其片段化。因此,在一些实施例中,片段化的基因组dna的大小可以选择为200-600bp范围内的dna片段。例如,片段化的基因组dna的大小可选择在50-750bp范围内、75-725bp范围内、100-700bp范围内、125-675bp范围内、150-650bp范围内、175-625bp范围内,或以下两种大小之间的任何范围:25bp、50bp、75bp、100bp、125bp、150bp、175bp、200bp、225bp、250bp、275bp、300bp、325bp、350bp、375bp、400bp、425bp、450bp、475bp、500bp、525bp、550bp、575bp、600bp、625bp、650bp、675bp、700bp、725bp、750bp、775bp、800bp或更大。片段化基因组dna的大小选择可由本领域中已知的任何方法进行,例如,使用琼脂糖凝胶电泳、固相可逆固定珠粒(例如ampurexp珠粒)、微流体仪器(例如caliperlabchipxt),可商购的库构建试剂盒(例如sagesciencepippinprep)等。片段化的基因组dna的大小选择可在获得片段化的基因组dna之后、在片段化的基因组dna被标记之后或在标记的、片段化的基因组dna被条形码化之后发生。因此,在一些实施例中,本公开提供用于测序单细胞基因组dna的方法,其包括从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化基因组dna的固化的微凝胶群体,以及片段化纯化基因组dna,以提供包括片段化的基因组dna的固化的微凝胶群体。在一些实施例中,在片段化纯化基因组dna的步骤之前,将包括纯化基因组dna的固化的微凝胶群体重新包封。因此,在一些实施例中,本公开提供用于测序单细胞基因组dna的方法,其包括从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化基因组dna的固化的微凝胶群体,将包括纯化基因组dna的固化的微凝胶群体包封到液滴中以提供含有纯化的基因组dna的液滴群体,以及片段化纯化基因组dna以提供含有片段化的基因组dna的液滴群体。在一些实施例中,将包括纯化基因组dna的固化的微凝胶群体包封到液滴中以提供含有纯化的基因组dna的液滴群体,其包括用试剂包封固化微凝胶用于片段化和标记纯化基因组dna。在一些实施例中,基因组dna的片段化和标记同时发生,例如,在标记步骤中,并且用试剂包封固化微凝胶用于片段化和标记纯化基因组dna,其包括用标记试剂包封固化微凝胶,例如包括转座酶和转座子的复合物。例如,在一些实施例中,纯化的含基因组dna的液滴的群体的每个成员包括复合物,其包括转座酶和转座子。在一些实施例中,用于测序单细胞基因组dna的方法包括从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供固化的微凝胶群体,其包括纯化的基因组dna。在一些实施例中,纯化的基因组dna经受使纯化的基因组dna片段化的条件,以提供固化的微凝胶群体,其包括片段化的基因组dna。在一些实施例中,片段化的基因组dna任选地用共同的衔接子序列标记。在一些实施例中,基因组dna的片段化和标记同时发生。在一些实施例中,基因组dna的片段化可以使用以下来实现:(neb)、转座子插入(nextera)、非特异性dna核酸内切酶如dnasei,或使用dna修复酶在扩增和裂解期间并入改质碱,如在扩增和使用endov和尿嘧啶糖基化酶的特异性裂解期间的dutp并入。流体动力学剪切也可用于片段化dna。在一些实施例中,方法包括通过转座子插入使纯化的基因组dna片段化,例如使用tn5转座子、mu转座子或本领域已知的任何其它合适的转座子。在此类实施例中,方法包括使纯化的基因组dna与包括转座酶和转座子的复合物接触。在一些实施例中,复合物包括转座子,其包括衔接子序列。使纯化的基因组dna与复合物接触导致片段化的基因组dna包括衔接子序列。在某些实施例中,由于转座酶的二聚体性质,片段化的基因组dna作为大分子复合物保持完整并且继续保留在固化的微凝胶群体内。因此,获得了包括片段化的基因组dna的固化的微凝胶群体,所述基因组dna任选地包括共同的衔接子序列。因此,如本公开提供的测序单细胞基因组dna的实例方法包括:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化的基因组dna的固化的微凝胶群体;将包括纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;和将纯化的含基因组dna的液滴的群体内的纯化基因组dna片段化,以提供含有片段化的基因组dna的液滴群体。使片段化的基因组dna条形码化所公开的方法可包括条形码化固化的微凝胶群体的步骤,所述固化的微凝胶群体包括片段化的基因组dna,其任选地包括共同的衔接子序列。进行条形码化,使得每个单个单细胞的片段化基因组dna与识别条形码序列,例如单个独特条形码序列相关联。在一些实施例中,片段化的基因组dna的条形码化可以在单个步骤中进行,例如,通过使用转座酶并入条形码序列,或者在两个步骤中进行,其中条形码序列被添加到具有例如连接酶或重叠延伸pcr的片段化的基因组dna中。在一些实施例中,包括片段化的基因组dna的固化的微凝胶群体可以与条形码序列库合并在一起,其中条形码序列的库的每个识别条形码序列(或识别条形码序列的群体),例如每个独特条形码序列(独特条形码序列的群体)分别包封在液滴中。因此,在一些实施例中,测序单细胞基因组dna的方法包括将包括片段化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有片段化的基因组dna的液滴群体。然后可以将含有片段化的基因组dna的液滴群体与含有条形码序列的液滴的库合并,以使得每个含有片段化的基因组dna的液滴与含有识别条形码序列(或识别条形码序列的群体),例如独特条形码序列(或独特条形码序列的群体)的液滴合并。方法可以进一步包括使含有片段化的基因组dna和条形码序列的液滴的群体经受足以将条形码序列酶并入片段化基因组dna的条件。将条形码序列并入片段化基因组dna的一种方法是使用与衔接子序列和条形码序列互补的引物,使得片段化基因组dna和条形码的产物扩增子可以彼此退火,并且通过延伸反应如dna聚合,彼此延伸,产生双链产物,包括附接到条形码序列的片段化的基因组dna。替代地或另外地,扩增靶标的引物本身可以被条形码化,使得在退火并延伸到靶标上时,产生的扩增子具有并入其中的条形码序列。这可以施加多种扩增策略,包括用pcr进行特异性扩增或用例如多重置换扩增(mda)进行非特异性扩增。可用于将条形码附接到片段化的基因组dna的替代或另外的酶反应是连接,包括平端或粘性末端连接。在此方法中,dna条形码与片段化的基因组dna和连接酶一起温育,导致条形码与靶标的连接。片段化基因组dna的末端可以根据用于连接的需要通过许多技术进行修改,包括通过使用被连接酶或片段引入的衔接子,以更好地控制添加到分子末端的条形码的数量。将条形码添加到片段化基因组dna的另一种方法是直接用转座酶或酶的组合,例如非特异性核酸内切酶或非特异性核酸内切酶(例如,)和连接酶的组合将其引入。例如,在此方法中,可以合成与转座酶相容的条形码。然后转座酶可以将纯化的基因组dna片段化并将条形码添加到片段分子的末端,在一个反应中进行反应的所有步骤。也可以使用和连接酶的组合,其中用于将核酸片段化为适合测序的大小,并且连接酶用于将条形码附接到片段末端。因此,如本公开提供的测序单细胞基因组dna的实例方法包括:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化的基因组dna的固化的微凝胶群体;将包括纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;将纯化的含基因组dna的液滴的群体内的纯化基因组dna片段化,以提供含有片段化的基因组dna的含dna液滴群体;和在片段化的含基因组dna的群体中对片段化的基因组dna或其扩增产物进行条形码化,以提供含有条形码化的片段化的基因组dna的液滴群体。在一些实施例中,在获得含有条形码化的片段化的基因组dna的液滴群体后,破坏包括液滴的群体的乳液,并纯化条形码化的片段化的dna以提供纯化的条形码化的片段化的基因组dna。可以进行任选大小选择步骤以选择特定大小的纯化的条形码化的基因组dna片段,其允许使用现有的测序平台进行测序。关于液滴中条形码核酸的另外公开在国际专利申请公开第wo2016/126871号中提供,其公开以引用的方式并入本文中。通过malbac进行分子扩增和条形码化作为标记/片段化的替代方案,通过使微凝胶与微流体滴管中的扩增试剂同向流动,可以使水凝胶中纯化的单细胞基因组以液滴进行malbac(多重退火和基于环的扩增循环)扩增反应。malbac反应通常在zong等人《全基因组检测单个人类细胞的单核苷酸和拷贝数变体(genome-widedetectionofsingle-nucleotideandcopy-numbervariationsofasinglehumancell)》,《科学(science)》,2012中描述,其公开以引用的方式并入本文中。简而言之,在malbac反应中,简并至基因组dna的引物退火并延伸。在第2周期和后期,在延伸和变性后形成发夹环。这些发夹不参与后面的反应循环,因为其处于环状构象中。在这种“准线性”扩增(6-10+循环)之后,使用单一引物的pcr以指数方式(10+循环)扩增环状产物。初始准线性反应的产物是环状扩增子。这些环可以通过(1)与条形码化的dna片段融合或(2)直接通过在malbac引物中插入条形码序列来进行条形码化。这里,为清楚起见,分别描述了两种新颖的条形码化方法。方法1:通过soe-pcr融合液滴中的分子条形码malbac反应使用单一引物(来自zong等人,2012):5'-gtgagtgatggttgaggtagtgtggagnnnnnnnn-3'(seqidno:1)27bp手柄8n代表性引物具有27bp手柄和8个简并碱基,但是其它变体也是可能的。然后,对在微凝胶和malbac扩增混合物中含有纯化dna的液滴进行热循环,例如,根据以下方案:液滴中的扩增子具有以下结构。注意,环是单链的,而27bp手柄区域是双链的:5'-gtgagtgatggttgaggtagtgtggag(seqidno:2)-环3'-ctccacactacctcaaccatcactcac(seqidno:3)-环单独地,条形码通过微滴中的数字pcr制得(例如,如本文所述)。双链条形码可具有以下一般结构:5'-pcr_handle-15n-gat-3'3'-pcr_handle*-15n*-gat*-5'pcr反应可以是非对称的,以通过使用过量的pcr_handle引物(例如,与限制性引物相比以1至5、10、20等比率)促进上单链的产生。pcr_handle引物也可以用生物素(或其它生物分子)功能化以促进使用抗生蛋白链菌素涂布的珠粒的下游捕获的条形码化的dna片段。数字条形码液滴与含有单细胞malbac扩增子的液滴微流体合并,例如以1∶1的比率。单循环的pcr用于退火并延伸条形码片段到使用其互补重叠区域的malbac扩增子上(图17)。替代地,可以进行多个pcr循环,并且使用位于条形码化的链的5′末端处的引物以液滴扩增条形码化的基因组dna(图18)。在此变体中,剩余的malbac引物可以用单链核酸外切酶(例如核酸外切酶i或类似物)消化。可以修饰用于pcr扩增的引物(例如通过3′硫代磷酸酯键或类似物)以防止核酸外切酶的降解。用于单循环延伸的实例pcr方案:95-1分钟60-30秒72-5分钟12-暂停用于延伸和指数扩增的实例pcr方案:破坏液滴并通过pcr富集条形码化的片段(在液滴中单循环延伸的情况下)。使用例如spri珠粒、凝胶电泳等的大小选择用于从扩增产物中去除单链dna条形码和引物二聚体。然后,可以根据测序平台的规格进行用于下一代测序的下游库制备。对于illumina合成测序(sbs)化学,条形码化的dsdna被片段化(酶,机械等),添加衔接子(通过连接,tn5[nextera]转座等),并通过pcr扩增库并测序。方法2:malbac引物的直接条形码化条形码化的malbac反应使用引物的库(由zong等人,2012年修改):5′-gtgagtgatggttgaggtagtgtggag[barcode]nnnnnnnn-3′(seqidno:4)27bp手柄8n将预定义的条形码寡核苷酸(4-12+bp条形码区域)在液滴中乳化以产生微滴的库,每个微滴含义微摩尔标度浓度的单一引物变体。当作为malbac扩增子环化时,引物形成组合条形码化的构建体(图19)。然后将微凝胶中的纯化dna与malbac扩增试剂和两个含引物的液滴微流体合并(图20)。使用与方法1相同的热循环方案进行malbac反应。得到的环状扩增子具有以下结构:[条形码1]-环5'-gtgagtgatggttgaggtagtgtggag-(seqidno:2)3'-ctccacactacctcaaccatcactcac-(seqidno:3)[条形码2]-环条形码序列1和2形成用于malbac环的独特组合标识符。在液滴中热循环后,破坏乳液并进行pcr以扩增条形码扩增子并产生双链条形码化的dna库。用于指数扩增的引物也可含有用于样品多路复用的条形码。用于ngs的准备取决于平台。然而,因为条形码是组合的并且位于构建体的5'和3'末端,所以在库制备步骤中dsdna片段不能被片段化。产生单细胞基因组测序读段的数据库本文描述的方法可包括对纯化的条形码化的片段化的基因组dna进行测序的步骤。dna序列可以通过可商购的下一代测序(ngs)平台实现,包括通过合成进行测序、通过连接测序、焦磷酸测序、使用可逆终止子化学、使用磷酸化荧光核苷酸或实时测序的平台。例如,纯化的条形码化的片段化的基因组dna可以使用定制的索引引物在illuminamiseq平台上测序。因此,如本公开提供的测序单细胞基因组dna的实例方法包括:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包括纯化的基因组dna的固化的微凝胶群体;将包括纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;将纯化的含基因组dna的液滴的群体内的纯化基因组dna片段化,以提供含有片段化的基因组dna的液滴群体;在片段化的含基因组dna的群体中对片段化的基因组dna或其扩增产物进行条形码化,以提供含有条形码化的片段化的基因组dna的液滴群体;从含有条形码化的片段化的基因组dna的液滴纯化条形码化的片段化的基因组dna,以提供纯化的条形码化的片段化的基因组dna;和对纯化的条形码化的片段化的基因组dna进行测序。从商业ngs平台获得的原始测序读段可以通过质量过滤并使用本领域已知的任何合适的脚本(例如,pythonscriptbarcodecleanup.py)通过条形码序列分组。在一些实施例中,如果超过约20%的碱基具有小于q20的质量得分(q-得分),则可丢弃给定的测序读段,表明碱基调用准确度为约99%。在一些实施例中,如果超过约5%、约10%、约15%、约20%、约25%、约30%具有小于q10、q20、q30、q40、q50、q60或更高的q-得分,则可丢弃给定的测序读段,表明碱基调用准确度分别为约90%、约99%、约99.9%、约99.99%、约99.999%、约99.9999%或更多。在一些实施例中,可以丢弃与含有小于50个读段的条形码相关的所有测序读段,以确保表示单细胞的所有条形码组含有足够数量的高质量读段。在一些实施例中,可以丢弃与含有小于30、小于40、小于50、小于60、小于70、小于80、小于90、小于100或更多的条形码相关的所有测序读段以确保表示单细胞的条形码组的质量。一旦原始测序读段按质量过滤并按条形码序列分组,序列可以输出到表,例如,关系数据库中的表,所述关系数据库例如sqlite数据库,包括与识别从单细胞获得的测序读段相关的领域。在一些实施例中,可以将序列输出到关系数据库中的表,例如,包括含有条形码序列、条形码组大小、唯一读段id号和读段序列的领域的sqlite数据库。在一些实施例中,例如,在分析合成细胞群体的情况下(参见实验部分),可以使用任何合适的可用软件程序(例如,bowtie2v2.2.9)对齐测序读段,并且sqlite表可通过用于每个测序读段的相关对齐信息被更新。在一些实施例中,例如,当分析环境样品时(参见实验部分),可以使用任何合适的可用软件程序(例如,krakenv0.10.5)通过分类法对测序读段进行分类,并且sqlite表可通过用于每个测序读段的相关分类信息被更新。在一些实施例中,可以使用任何合适的可用脚本,例如pythonscriptpurity.py,从参考对齐数据或系统发育标记计算条形码组纯度。效用本公开提供了对来自单细胞群体的单细胞基因组dna进行测序的方法。本文提供的方法发现可用于癌症的拷贝数变体分析。例如,在癌症中,细胞经历快速进化,导致其基因组的大量突变。一种形式的突变是拷贝数变体,其中基因组的不同部分被复制或擦除。在一些实施例中,本文提供的方法允许跨基因组计数已知序列。使用本文提供的方法的优点是拷贝数变体分析可以在基因组的低覆盖度下进行,通常小于1%,允许其在不必进行大量测序的情况下进行测量。在一些实施例中,本文提供的方法发现可用于血液测序,其中血液测序包括测序血液样品(例如免疫细胞)中所有所关注的细胞。例如,可以收集血液样品并萃取相关的细胞类型。免疫细胞对循环系统进行采样,并且因此,其生物状态可以表示疾病,例如感染、败血症、癌症、自身免疫性疾病等。在一些实施例中,对来自血液样品的免疫细胞的核酸进行测序可以允许检测各种患病状态。在一些实施例中,可以使用本文提供的方法检测稀有细胞,如循环肿瘤细胞和循环胚胎细胞。在一些实施例中,尽管来自含有循环肿瘤和/或胚胎细胞的血液样品的大多数测序细胞将不对应于所关注的细胞群体,但是当任何此类循环肿瘤和/或胚胎细胞被识别时,关于其基因组的完整信息可以回收。在一些实施例中,本文提供的方法发现可用于宏基因组学领域。例如,本文提供的方法发现可用于研究多样的微生物系统,其中其分析在在识别稀有系统成员和使其被回收用于具体研究,例如回收其核酸序列中可以是有价值的。在一些实施例中,本文提供的方法发现可用于研究潜在的人类免疫缺陷病毒(hiv)感染。使用如本文所述的方法,可以基于hiv基因的存在对来自感染个体的细胞进行分选。可以对分选的细胞进行测序以回收关于其基因组如何被病毒调节的信息。本领域技术人员将能够设想使用本文所述方法的能力,并且能够使这些方法适用于任何应用。虽然本文提供的方法主要是针对细胞基因组dna进行描述,但应注意,sic-seq方法还提供了分离和条形码大dna分子的方法,无论其起源于何种实体。虽然所描述的方法集中于细胞,但是类似的方法可施加到其基因组可在凝胶基质内被捕获和处理的病毒上。因此,例如,在一些实施例中,本公开提供了测序基因组dna(例如病毒基因组dna)的方法,所述方法包括将生物实体群体(例如病毒)包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个生物实体;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的生物实体的裂解条件;从含于大量固化的微凝胶群体内的生物实体纯化基因组dna,以提供包含纯化的基因组dna的固化的微凝胶群体;将包含纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;将纯化的含基因组dna的液滴的群体内的纯化基因组dna片段化,以提供含有片段化的基因组dna的液滴群体;在片段化的含基因组dna的群体中对片段化的基因组dna或其扩增产物进行条形码化,以提供含有条形码化的片段化的基因组dna的液滴群体;从含有条形码化的片段化的基因组dna的液滴纯化条形码化的片段化的基因组dna,以提供纯化的条形码化的片段化的基因组dna;和对纯化的条形码化的片段化的基因组dna进行测序。类似地,应当理解,如下文所提供本公开第1号至第36号的每个非限制性方面可被修改为指合适的生物实体,例如病毒而非细胞。可根据需要修改每个本文所描述的方法以与基于非细胞的生物实体或大dna分子一起使用。装置和系统如上所述,所公开主题的实施例使用包括微流体装置的系统和/或装置。本公开的装置包括以上结合本发明方法描述的所有装置。本公开的微流体装置可以以各种方式表征。在一些方面,例如,提供微流体系统和/或装置,其包括一个或多个液滴制造器,配置成产生如本文所述的液滴和/或一个或多个流动通道。在一些方面,一个或多个流动通道可操作地连接(例如,流体连接)到一个或多个液滴制造器和/或配置成从其接收一个或多个液滴。如本文所使用的,“可操作地连接”和“可操作地耦合”意指以特定方式(例如,以允许流体,例如水,移动和/或电力传输的方式)连接,这允许公开系统或装置及其各种组件以本文所述的方式有效地操作。如上所述,微流体装置可包括一个或多个流动通道,例如,液滴可以进入、流出和/或通过的流动通道。在某些实施例中,流动通道是一个或多个“微”通道。这样的通道可具有至少一个约一毫米或更小(例如,小于或等于约1毫米)的横截面尺寸。对于某些应用,可以调节此尺寸;在一些实施例中,至少一个横截面尺寸为约500微米或更小。在一些实施例中,横截面尺寸为约100微米或更小,或约10微米或更小,且有时约1微米或更小。横断面尺寸是通常垂直于中心线流动方向的尺寸,但应该理解的是,当遇到流过弯头或倾向于改变流动方向的其它特征时,场景中的横截面尺寸不必严格垂直于流动。还应理解,在一些实施例中,微通道可以具有两个或更多个横截面尺寸,如矩形横截面的高度和宽度或椭圆形横截面的长轴和短轴。可以将这些尺寸中的任何一个与这里给出的尺寸进行比较。注意,本公开中使用的微通道可具有两个严重不成比例的尺寸-例如,具有约100-200微米的高度的矩形横截面和约1厘米或更大的宽度。当然,某些装置可使用其中两个或更多个轴在大小上非常相似或甚至相同的通道(例如,具有正方形或环形横截面的通道)。在本公开的一些实施例中,微流体装置使用微制造技术制造。可以使用这种技术来制造集成电路(ic)、微机电装置(mems)、显示装置等。可用于在微流体装置制造中产生小尺寸图案的微型制造工艺类型是光刻(包括x射线光刻、电子束光刻等)、自对齐沉积和蚀刻技术、各向异性沉积和蚀刻工艺、自组装掩模形成(例如,形成疏水-亲水共聚物层)等。鉴于以上所述,应该理解的是,本文描述的一些原理和设计特征可以扩展到更大的装置和系统,包括使用达到毫米或甚至厘米标度通道横截面的通道的装置和系统。因此,当将一些装置和系统描述为“微流体”时,希望是在某些实施例中,所述描述同样施加到一些较大规模的装置。当提及微流体“装置”时,其通常旨在表示单个实体,其中一个或多个通道、贮存器、客户端等共享连续衬底,其可以是单片或不是单片。微流体装置的方面包括存在一个或多个流体流动路径,例如通道,具有如本文所述的尺寸。微流体“系统”可包括一个或多个微流体装置和相关的流体连接、电连接、控制/逻辑特征等。系统还可以包括以下中的一个或多个:(a)温度控制模块,用于控制本发明装置的一个或多个部分和/或其中的液滴的温度,并且可操作地连接到微流体装置,(b)检测装置,即可操作地连接到微流体装置的检测器,例如光学成像器,(c)可操作地连接到微流体装置的培养箱,例如细胞培养箱,和(d)可操作地连接到微流体装置的测序仪。本发明系统还可包括一个或多个传送器,其配置成将衬底从第一液滴,接收位置移动(例如,传送)到(a)-(d)中的一个或多个。本发明装置和系统包括用于将液滴分选到一个或多个流动通道中的一个或多个分选器。这种分选器可以基于液滴的一个或多个特征来分选和分配液滴,所述特征包括组成、大小、形状、浮力或其它特征。装置的各方面还包括一个或多个检测装置,即检测器,例如光学成像器,配置成用于检测一个或多个液滴的存在,或其一个或多个特征,包括其组成。在一些实施例中,检测装置配置成在一个或多个流动通道中识别一个或多个液滴的一种或多种组分。在各种实施例中,本公开的微流体装置提供流体介质的连续流动。流过微流体装置中的流过通道的流体表现出许多独特特性。通常,无量纲雷诺数极低,导致流动始终保持层流状态。此外,在此方案中,两种流体连接将不容易混合,并且单独的扩散可以驱动两种化合物的混合。另外,在一些实施例中,本发明装置包括一个或多个温度和/或压力控制模块。这种模块可以能够调节装置的一个或多个流动通道中的载体流体的温度和/或压力。更具体地,温度控制模块可以是一个或多个热循环仪。参考图1,图区a、b和c描述示例性实施例,其描绘了可用于以下的微流体装置的示意图:a)产生条形码液滴和包封在微凝胶(例如琼脂糖微凝胶)中的细胞,b)用片段化和/或标记试剂,例如标记试剂重新包封微凝胶,和c)将微凝胶液滴与条形码液滴和pcr液滴合并。如图1,图区a所示,单细胞被包装成熔融凝胶液滴,并被载体流体(例如油)包封和间隔以提供包封的单细胞。由于混合物的粘度和界面张力特性,熔融凝胶液滴形成可能是缓慢的。为了加速熔融凝胶液滴的形成,可以使用气泡触发(abate,a.r.和weitz,d.a.,labchip,11(10):1713-1716,2011),其公开以引用的方式并入本文中。在一些实施例中,熔融凝胶溶液和细胞在喷射流动条件下同向流入液滴发生器。在喷射流动条件下也引入油,使得射流由油相中的水相形成。气泡触发通过作为气泡注入或通过与喷射一起注入气流,在喷射器附近引入气泡。在一些情况下,气泡会扰乱喷射器,导致其破裂成液滴。如果气泡周期性地间隔开,则在均匀大小的气泡之间形成液滴,从而增加单分散液滴产生的速率。因此,在一些实施例中,本公开内容提供了一种系统,其包括微流体装置、熔融凝胶贮存器和加热元件,所述微流体装置包括同向流动液滴制造器,所述同向流动液滴制造器包括:第一输入通道,其被配置成向流动通道提供多个细胞;第二输入通道,其被配置成从凝胶贮存器向流动通道提供熔融凝胶流,其中加热元件定位在凝胶贮存器附近并且被配置成向凝胶贮存器施加足够的热量以使熔融凝胶在熔融凝胶贮存器中保持呈熔融状态;以及第三输入通道和第四输入通道,其定位在流动通道的相对侧以及第一和第二输入通道的下游,其中第三和第四输入通道被配置成向流动通道提供不混溶相流体流。现在参考图1,图区b,可用于进行本公开的方法的装置包括用于利用片段化和/或标记试剂(例如标记试剂)重新包封微凝胶的微流体装置。如图1,图区b所示,微凝胶与片段化和/或标记试剂(例如标记试剂)组合,并被载体流体(例如油)包封和间隔以提供包封的单细胞。现在参考图1,图区c,可用于进行本公开的方法的装置还可进一步包括所提供的用于并入试剂(例如,pcr试剂、细胞裂解试剂等)和用于并入条形码核酸序列的贮存器,其中液体电极、深沟、液滴合以及用于间隔所识别的载体流体,例如油、条形码液滴和pcr液滴的贮存器。因此,在一些实施例中,本公开提供了一种装置,其包括用于以下的组分:(a)将两个或更多个液滴的群体引入到流动通道中,(i)其中流动通道包括与一个或多个电极或一个或多个电极的一个或多个部分相关的液滴合并部分,所述电极被配置成在流动通道的液滴合并部分中施加电场,(ii)其中两个或更多个液滴的群体在分别来自两个或更多个单独入口通道的单个接合处被引入到流动通道,和(iii)其中两个或更多个液滴的群体被引入到流动通道,使得来自每个入口通道的液滴输入由于流体动力学效应而至少部分地同步,导致液滴的间隔组的喷射,其中至少一些液滴的间隔组包括来自两个或更多个液滴的群体中的每个的液滴;(b)将液滴的间隔组流入液滴合并部分;以及(c)通过使用一个或多个电极或一个或多个电极的一个或多个部分在流动通道的液滴合并部分中施加电场,以在间隔组内合并液滴。本公开的示例性非限制性方面上述本主题的各方面(包括实施例)可以单独或与一个或多个其它方面或实施例组合有益。在不限制前述描述的情况下,下面提供本公开的某些非限制性方面(组a和组b)。对于本领域普通技术人员在阅读本公开时将显而易见的是,每个单独编号的方面可以与前面或后面的任何单独编号的方面一起使用或组合。这旨在为所有这些方面的组合提供支持,并且不限于以下明确提供的方面的组合:组a1.一种对单细胞基因组dna进行测序的方法,所述方法包含:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包含纯化的基因组dna的固化的微凝胶群体;将包含纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;将纯化的含基因组dna的液滴的群体内的纯化基因组dna片段化,以提供含有片段化的基因组dna的液滴群体;在片段化的含基因组dna的群体中对片段化的基因组dna或其扩增产物进行条形码化,以提供含有条形码化的片段化的基因组dna的液滴群体;从含有条形码化的片段化的基因组dna的液滴纯化条形码化的片段化的基因组dna,以提供纯化的条形码化的片段化的基因组dna;和对纯化的条形码化的片段化的基因组dna进行测序。2.根据1的方法,其中条形码包含将每个含有片段化的基因组dna的液滴与含有条形码的液滴合并。3.根据2的方法,其中每个含有条形码的液滴包含独特的核酸条形码序列。4.根据1至3中任一项的方法,其中方法包含将衔接子核酸序列并入片段化的基因组dna。5.根据1至4中任一项的方法,其中单细胞的群体包含真核细胞。6.根据5的方法,其中单细胞的群体包含哺乳动物细胞。7.根据1至4中任一项的方法,其中单细胞的群体包含细菌细胞。8.根据1至5中任一项的方法,其中单细胞的群体包含真菌细胞。9.根据1至8中任一项的方法,其中熔融凝胶液滴包含水凝胶聚合物。10.根据9的方法,其中水凝胶聚合物包含热响应性聚合物。11.根据10的方法,其中热响应性聚合物是琼脂糖。12.根据1至11中任一项的方法,其中固化包含冷却熔融凝胶液滴的群体。13.根据1至9中任一项的方法,其中熔融凝胶液滴包含聚乙二醇(peg)。14.根据13的方法,其中固化包含使peg化学交联。15.根据13的方法,其中固化包含使peg光交联。16.根据1至9中任一项的方法,其中熔融凝胶液滴包含丙烯酰胺。17.根据16的方法,其中固化包含使丙烯酰胺化学交联。18.根据16的方法,其中固化包含使丙烯酰胺光交联。19.根据1至9中任一项的方法,其中熔融凝胶液滴包含藻酸盐。20.根据19的方法,其中固化包含向熔融凝胶液滴中添加钙。21.根据1至20中任一项的方法,其中固化微凝胶包含大小设定成将基因组dna保留在固化微凝胶内的孔。22.根据1至21中任一项的方法,其中将单细胞的群体包封在熔融凝胶液滴中的步骤包含添加油。23.根据1至22中任一项的方法,其中暴露包含使大量固化的微凝胶群体与裂解酶接触以裂解含于固化的微凝胶群体中的细胞。24.根据23的方法,其中裂解酶选自酵母裂解酶、溶葡球菌酶、变溶菌素、溶菌酶或其组合。25.根据1至24中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含使固化的微凝胶群体与清洁剂接触以溶解含于固化的微凝胶群体内的细胞物质。26.根据25的方法,其中清洁剂选自十二烷基硫酸锂、十二烷基硫酸钠或其组合。27.根据1至26中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含使固化的微凝胶群体与蛋白酶接触以消化含于固化的微凝胶群体内的细胞蛋白质。28.根据27的方法,其中蛋白酶是蛋白酶k。29.根据1至28中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含洗涤固化的微凝胶群体的步骤,其中洗涤固化的微凝胶群体的步骤包含使固化的微凝胶群体与洗涤缓冲液接触。30.根据1至29中任一项的方法,其中每个纯化的含基因组dna的液滴的群体包含复合物,其包含转座酶和转座子。31.根据1至30中任一项的方法,其中片段化的步骤包含使纯化的基因组dna与包含转座酶和转座子的复合物接触。32.根据31的方法,其中复合物包含转座子,其包含衔接子序列。33.根据32的方法,其中使纯化的核酸与复合物接触提供包含衔接子序列的片段化的基因组dna。34.根据1至33中任一项的方法,其中将单细胞群体包封在熔融凝胶液滴中的步骤和将片段化的基因组dna或其扩增产物条形码化的步骤使用微流体装置进行。35.根据1至34中任一项的方法,其中一种或多种步骤为固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包含纯化的基因组dna的固化的微凝胶群体;和将包含纯化的基因组dna的大量固化的微凝胶群体内的纯化基因组dna片段化以提供包含片段化的基因组dna的固化的微凝胶群体不使用微流体装置进行。36.根据1至35中任一项的方法,其中单细胞的群体是单细胞微生物的异质群体。37.一种系统,所述系统包含微流体装置、熔融凝胶贮存器和加热元件,所述微流体装置包含同向流动液滴制造器,其包含第一输入通道,其被配置成向流动通道提供多个细胞,第二输入通道,其被配置成从凝胶贮存器向流动通道提供熔融凝胶流,其中加热元件定位在凝胶贮存器附近并且被配置成向凝胶贮存器施加足够的热量以使熔融凝胶在熔融凝胶贮存器中保持呈熔融状态,以及第三输入通道和第四输入通道,其定位在流动通道的相对侧以及第一和第二输入通道的下游,其中第三和第四输入通道被配置成向流动通道提供不混溶相流体流。组b1.一种测序单细胞基因组dna的方法,所述方法包含:将单细胞群体包封在熔融凝胶液滴中以提供熔融凝胶液滴群体,其中群体的每个熔融凝胶液滴含有零个或一个细胞;固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包含纯化的基因组dna的固化的微凝胶群体;将包含纯化的基因组dna的固化的微凝胶群体包封到液滴中,以提供含有纯化的基因组dna的液滴群体;对基因组dna或其一种或多种扩增产物进行条形码化,以提供含有条形码化的基因组dna的液滴群体;从条形码化的含基因组dna的液滴纯化条形码化的基因组dna,以提供纯化的条形码化的基因组dna;和对纯化的条形码化的基因组dna进行测序。2.根据1的方法,其中条形码化包含将每个纯化的含基因组dna的液滴与含有条形码的液滴合并。3.根据2的方法,其中每个含有条形码的液滴包含独特的核酸条形码序列。4.根据1至3中任一项的方法,包含在纯化的含基因组dna的液滴中进行malbac扩增反应,例如,如本文所述,关于通过malbac的分子扩增和条形码化。5.根据1至4中任一项的方法,其中单细胞的群体包含真核细胞。6.根据5的方法,其中单细胞的群体包含哺乳动物细胞。7.根据1至4中任一项的方法,其中单细胞的群体包含细菌细胞。8.根据1至5中任一项的方法,其中单细胞的群体包含真菌细胞。9.根据1至8中任一项的方法,其中熔融凝胶液滴包含水凝胶聚合物。10.根据9的方法,其中水凝胶聚合物包含热响应性聚合物。11.根据10的方法,其中热响应性聚合物是琼脂糖。12.根据1至11中任一项的方法,其中固化包含冷却熔融凝胶液滴的群体。13.根据1至9中任一项的方法,其中熔融凝胶液滴包含聚乙二醇(peg)。14.根据13的方法,其中固化包含使peg化学交联。15.根据13的方法,其中固化包含使peg光交联。16.根据1至9中任一项的方法,其中熔融凝胶液滴包含丙烯酰胺。17.根据16的方法,其中固化包含使丙烯酰胺化学交联。18.根据16的方法,其中固化包含使丙烯酰胺光交联。19.根据1至9中任一项的方法,其中熔融凝胶液滴包含藻酸盐。20.根据19的方法,其中固化包含向熔融凝胶液滴中添加钙。21.根据1至20中任一项的方法,其中固化微凝胶包含大小设定成将基因组dna保留在固化微凝胶内的孔。22.根据1至21中任一项的方法,其中将单细胞的群体包封在熔融凝胶液滴中的步骤包含添加油。23.根据1至22中任一项的方法,其中暴露包含使大量固化的微凝胶群体与裂解酶接触以裂解含于固化的微凝胶群体中的细胞。24.根据23的方法,其中裂解酶选自酵母裂解酶、溶葡球菌酶、变溶菌素、溶菌酶或其组合。25.根据1至24中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含使固化的微凝胶群体与清洁剂接触以溶解含于固化的微凝胶群体内的细胞物质。26.根据25的方法,其中清洁剂选自十二烷基硫酸锂、十二烷基硫酸钠或其组合。27.根据1至26中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含使固化的微凝胶群体与蛋白酶接触以消化含于固化的微凝胶群体内的细胞蛋白质。28.根据27的方法,其中蛋白酶是蛋白酶k。29.根据1至28中任一项的方法,其中从含于固化的微凝胶群体内的细胞纯化基因组dna的步骤包含洗涤固化的微凝胶群体的步骤,其中洗涤固化的微凝胶群体的步骤包含使固化的微凝胶群体与洗涤缓冲液接触。30.根据1至29中任一项的方法,其中将单细胞群体包封在熔融凝胶液滴中的步骤和将片段化的基因组dna或其扩增产物条形码化的步骤使用微流体装置进行。31.根据1至30中任一项的方法,其中一种或多种步骤为固化熔融凝胶液滴的群体以提供固化的微凝胶群体液滴;破坏固化微凝胶液滴的乳液以提供固化的微凝胶群体;将大量固化的微凝胶群体暴露于足以裂解含于固化的微凝胶群体内的细胞的裂解条件;和从含于大量固化的微凝胶群体内的细胞纯化基因组dna,以提供包含纯化的基因组dna的固化的微凝胶群体不使用微流体装置进行。32.根据1至31中任一项的方法,其中单细胞的群体是单细胞微生物的异质群体。本领域普通技术人员显而易见的是可在不脱离本发明的精神和范围的情况下作出各种改变和修改。实例提出以下实例以便向本领域普通技术人员完整地公开且描述如何制造和使用本发明,并且并不旨在限制本发明的范围,也不旨在表示下文实验为所进行的全部或唯一实验。已经努力确保关于所用数字(例如数量、温度等)的准确度,但应该考虑到一些实验误差和偏差。除非另外指明,否则份数是重量份,分子量是重量平均分子量,温度是摄氏度,并且压力是或接近大气压。本说明书中所引用的所有公开案和专利申请均以引用的方式并入本文中,就如同各个别公开案或专利申请专门且单独地指出以引用的方式并入一般。已经根据发现或提出的包含用于实践本发明的优选模式的特定实施例描述了本发明。本领域的普通技术人员应了解,根据本公开,在不脱离本发明的预定范围的情况下,可以对示例的特定实施例作出多种修改和改变。希望所有此类修改包括于所附权利要求书的范围内。实例1:使用液滴微流体条形码进行超高通量单细胞基因组测序。本公开提供了超高通量单细胞基因组测序(sic-seq),一种能够每次运行测序>50,000个单细胞基因组的液滴微流体方法。方法通过对在对照比例下的含有已知物种的微生物的人工群体进行测序来验证,获得每个细胞约0.1%的平均覆盖度、均匀的基因组取样和物种比例的准确估计。此外,sic-seq产生宏基因组数据库,其中读段按单细胞分组。反过来,此数据库实现了类似于常规流式细胞术的新型“计算机模拟血细胞计数”,除了基于基因组序列标记在计算上出现的所有分选,并且不需要指定这些生物标记物以先验地收集数据。为了证明这一点,将sic-seq和计算机模拟血细胞计数施加到海洋微生物样品,并用于测量抗生素抗性基因、毒力因子和噬菌体相关序列如何在整个群体中分布。在不必进行另外的湿实验室实验的情况下重复对基因组群体进行分选的能力使得通过假设快速迭代并且增强可以通过所学内容发现的内容。其对于产生特征序列之间的相关性图、推断不同表型在单细胞内的相关性以及基因元件如何通过群落传播是有价值的。材料和方法:微流体装置:为了制造微流体装置,将聚(二甲基硅氧烷)(道康宁(dowcorning),sylgard184)倾倒在使用uv光刻在硅晶片(universitywafer)上图案化的负光致抗蚀剂(microchem,目录号su-83025)上。将pdms装置在烘箱中固化1小时,用金属刮刀萃取,并用0.75mm活检芯(世界精确度仪器(worldprecisioninstruments),目录号504529)打孔以产生入口和出口。使用氧等离子体清洁剂(harrickplasma)和用aquapel(ppgindustries)处理的通道将装置粘合到玻璃载片上,并在80℃下烘烤10分钟以使其疏水。条形码化乳液:通过数字pcr方法制备条形码化乳液,其中条形码化寡核苷酸在含有pcr试剂的液滴中作为单分子扩增。将在0.01pm浓度下的条形码化寡核苷酸(gcagctggcgtaatagcgagtacaatctgctctgatgccgcatagnnnnnnnnnnnnnnntaagccagccccgacact)(seqidno:5)(idt)添加到含有以下的pcr反应混合物中:1xneb福讯热启动挠曲主混合物(phusionhotstartflexmastermix)(neb,目录号m0536l)、2%(w/v)tween20(西格玛-阿尔德里奇(sigma-aldrich),目录号p9416)、5%(w/v)peg-6000(圣克鲁兹生物技术(santacruzbiotechnology),目录号sc-302016),和400nm引物fl128(ctgtctcttatacacatctccgagcccacgagacgtgtcggggctggctta)(seqidno:6)和fl129(caagcagaagacggcatacgagatcagctggcgtaatagcg(seqidno:7),含有p7衔接子序列)(idt)。将具有2%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂(008-氟表面活性剂,ran技术)的pcr混合物和hfe-7500氟化油(3m)装入单独的1ml注射器(bd)中并分别以300和500μl/hr注入使用由定制python脚本(“https:”之后“//github.”之后“com/abatelab/泵-控制-程序(pump-control-program)”)控制的注射泵(newera,目录号ne-501)的流动聚焦液滴制造器。将乳液收集在pcr管中,并通过移液管去除乳液下面的油,并用具有5%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂的fc-40氟化油(西格玛-阿尔德里奇,目录号51142-49-5)代替,以改善热稳定性。使用以下程序对乳液进行热循环(拜耳雷德(bio-rad),t100):98℃3分钟,之后进行40个循环,其中每秒2℃的升温速率为98℃10秒、62℃20秒,以及72℃20秒,之后在12℃下保持。使用hfe-7500油中的10xsybrgreeni(赛默飞世尔科技(thermofisherscientific))进行的荧光dna染色用于在荧光显微镜(生命技术(lifetechnologies),目录号amafd1000)下定量条形码化包封率。细胞培养和计数:产生一个用于验证sic-seq工作流程的人工群落,表皮葡萄球菌、酿酒酵母(菌株s288c),和枯草芽孢杆菌(菌株168)的液体培养物在振荡培养箱中生长过夜。使用以下培养条件:表皮葡萄球菌和枯草芽孢杆菌在37℃下在3mllb培养液中生长;酿酒酵母在30℃下在3mlypd培养液中生长。通过使用显微镜手动计数塑料载片(赛默飞世尔科技,目录号c10228)上的液体培养物的系列稀释液来确定细胞浓度。将培养物保持在4℃,然后用于微流体实验(参见标题为《琼脂糖滴中的细胞包封(cellencapsulationinagarosedroplets)》的部分)。水样收集和过滤:为了获得微生物群落的天然样品,从美国加利福尼亚州西旧金山(westsanfrancisco,california,usa)(37°44'55.6"n122°30'33.6"w)的oceanbeach收集海水。通过将两个1000ml玻璃瓶浸没在距离海岸线约20m的水面下,获得约2l的水。在运输到实验室期间将样品置于冰上。使100ml样品穿过40μm细胞过滤器(康宁,产品号352340)以去除大的碎片,包括沙子。将样品装入0.45μm真空过滤器(密理博(millipore),目录号schvu01re);此过滤步骤分离在膜上捕获的微生物和在滤液中丢弃的病毒。使用刮刀从设备中萃取膜并插入15ml离心管中,向其中添加5mlpbs。将管在高速下涡旋约2分钟以从膜中释放细菌细胞。最后,将细胞溶液装入10ml注射器中并穿过5μm注射器过滤器(密理博,目录号slsv025ls)以去除剩余的大颗粒。使用与液体培养物相同的方案计数海洋细胞。琼脂糖微凝胶中的细胞包封:为了通过sic-seq工作流程制备用于加工的人工群落,将冷冻的细胞原料(兹莫研究(zymoresearch),目录号d6300)在室温水浴中温和解冻。通过在显微镜下手动细胞计数测定细胞浓度,并稀释至适当浓度以进行单细胞包封。将计算的细胞溶液体积转移至1.5ml离心管(飞世尔科技公司(fisherscientific))中并在1mlpbs中洗涤两次。将细胞再悬于含有以下的1mlpbs溶液中:17%optiprep密度梯度培养基(西格玛-阿尔德里奇)、0.1mg/mlbsa(西格玛-阿尔德里奇,目录号a9418)和1%(v/v)普朗尼克(pluronic)f-68(生命技术(lifetechnologies))。将细胞溶液装入1ml注射器中并置于注射泵(newera,目录号ne-501)上。在1.5ml离心管中制备1ml3%低凝胶温度琼脂糖溶液(西格玛-阿尔德里奇,目录号a9414)和te缓冲液(天惠华(teknova),目录号t0225),并在90℃下在嵌段上加热约10分钟以完全溶解琼脂糖粉末。将热琼脂糖转移至1ml注射器中并置于注射泵上。为了在微流体实验期间保持琼脂糖熔融,将个人空间加热器放置在离琼脂糖注射器约5cm处并设定为在高温下连续运行。将具有2%(w/w)去质子化的krytox表面活性剂(杜邦(dupont,目录号157fsh)的hfe-7500氟化油装入3ml注射器中。将细胞溶液、熔融琼脂糖和油分别以200、200和400μl/hr的流动速率注入同向流动液滴制造器中,以形成1.5%琼脂糖微凝胶。约500μl的液滴收集在冰上的15ml离心管中并在4℃下温育30分钟,以确保微凝胶的完全固化。在水性缓冲液中再悬微量凝胶:将液滴以300g离心1分钟,以最大化的将乳液与油的分离。使用5ml注射器从管中萃取油层并丢弃。使用hfe-7500中的2ml10%(v/v)全氟辛醇(西格玛-阿尔德里奇,目录号370533)溶液破坏乳液;然后通过移液将乳液混合并在300g下离心1分钟。使用注射器从管中去除油,并重复液滴破坏步骤。在液滴破坏后,将含有1%(v/v)span80(西格玛-阿尔德里奇)的2ml己烷添加到微凝胶中以溶解任何剩余的油,并将此溶液混合并在300g下离心1分钟。从管中去除己烷上清液,并重复己烷添加步骤。最后,将微凝胶在含有0.1%(v/v)tritonx-100非离子表面活性剂(西格玛-阿尔德里奇)的10ml水性溶液te缓冲液中洗涤三次。将微凝胶在1000g下离心2分钟,并在洗涤之间抽吸上清液。在细胞裂解之前,将洗过的微凝胶储存在4℃下的5mlte缓冲液中。微凝胶中的细胞裂解:为了裂解微凝胶中的细胞,将颗粒浸没在含有以下的2mlte缓冲溶液的溶液中:10mmdtt(manu)、2.5mmedta(天惠华(teknova))和10mmnacl(西格玛-阿尔德里奇)。还包括以下量的裂解酶:4u酵母裂解酶(兹莫研究)、10u溶葡球菌素(西格玛-阿尔德里奇,目录号l7386)、100u变溶菌素(西格玛-阿尔德里奇,目录号m9901)和40mg溶菌酶(mp生物医学(mpbiomedicals),目录号195303)。细胞裂解在37℃下的振荡培养箱中过夜进行。将混浊的裂解物混合物在1000g下离心1分钟,去除上清液,并添加3ml的含有0.5%(w/v)十二烷基硫酸锂(西格玛-阿尔德里奇)和te缓冲液中的10mmedta的溶液,随着4u蛋白酶k(neb)溶解细胞碎片并消化细胞蛋白。将溶液在50℃下在加热块上温育30分钟。裂解后,彻底洗涤微凝胶以确保完全去除可能抑制下游分子生物学反应的清洁剂和其它化学物种。在添加洗涤溶液之间在1000g的离心幅度下以10ml体积发生以下洗涤:用2%(v/v)tween20在水中洗涤一次;在100%乙醇(koptec)中洗涤一次以使任何剩余的蛋白酶k变性;以及用0.02%(v/v)tween20在水中洗涤五次。微凝胶中基因组dna的标记:使用来自nexteradna库制备试剂盒(illumina,目录号fc-121-1030)的试剂,将含有高分子量基因组dna的经洗涤和裂解的凝胶同时片段化并用通用衔接子序列标记。将微凝胶重新包封到液滴中以最小化标记步骤期间的交叉污染。制备192μldi水、200μl标记缓冲液和8μlnextera酶的溶液并装入1ml注射器中。将微凝胶和标记溶液注入重新包封装置(图1,图区b)。将重新包封的微凝胶在50℃下在加热块上的1.5ml管中温育1小时。包封细胞的微流体条形码:将标记的微凝胶、条形码化液滴和500μlpcr溶液,其含有:1xinvitrogen铂多重pcr主混合物(赛默飞世尔科技,目录号4464268)、400nm引物fl127(aatgatacggcgaccaccgagatctacactcgtcggcagcgtc(seqidno:8),containsp5adaptersequence)和fl129(caagcagaagacggcatacgagatcagctggcgtaatagcg)(seqidno:7),来自nexteraxt试剂盒(0.2%sds)(illumina,目录号fc-131-1024)的50x稀释的nt缓冲液、1%(w/v)tween20、1%(w/v)peg-6000、2.5u/μlbst2.0warmstartdna聚合酶(neb,目录号m0538s)各自装入1ml注射器中并注入合并装置,如图1中所示。使用具有2%(w/w)008-含氟表面活性剂的hfe-7500氟化油作为乳液的连续相。使用连接到冷阴极荧光逆变器和dc电源(mastech)的电极实现条形码和凝胶液滴乳液的合并。电源下的2.0v电压在电极处产生约2kv的ac电势,这致使触摸液滴合并。将乳液收集在0.5ml薄壁pcr管(施加生物科学(appliedbiosciences))中,并在热循环之前用5%(w/w)008-含氟表面活性剂替换为具有fc-40的hfe-7500,其具有以下方案:65℃5分钟,95℃2分钟,然后30个循环,2℃/s下升温速率为95℃15秒、60℃1分钟、72℃1分钟,并且然后72℃5分钟,其中任选12℃保持过夜。在热循环后,使用微量移液管去除大的(聚结的)液滴,并通过添加20μl全氟辛醇并在微型离心机中短暂离心来破坏乳液。收集上层水相,并使用兹莫dna清洁&浓缩器-5试剂盒(zymodnaclean&concentrator-5kit)(兹莫研究)纯化dna库。库是在200-600bp范围内使用以下针对dna片段进行的大小-选择:使用agencourtampurexp珠粒(贝克曼库尔特公司(beckmancoulter)),使用生物分析仪2100仪器和高灵敏度dna芯片(安捷伦(agilent))定量,并使用定制指数引物(fl166)在illuminamiseq上测序。产生sic读段数据库:miseq产生的fastq文件的原始读段按质量过滤,并使用python脚本barcodecleanup.py按条形码序列分组。如果超过20%的碱基的q-得分低于q20,则丢弃给定的读段,并丢弃与含有少于50个读段的条形码相关的所有读段。此步骤确保表示单细胞的所有条形码组含有足够数量的高质量读段。将得到的读段输出到sqlite数据库中的表中,其含有条形码序列、条形码组大小、唯一读段id号和读段序列的领域。当参考基因组已知时,如在合成细胞群体实验的情况下,使用具有默认设置的bowtie2v2.2.9对齐读段,并且使用每次读段的相关对齐信息更新sqlite表。对于环境样品,读段按照分类法使用krakenv0.10.5进行分类,并设置“--quick--min-hits2”选项,并将输出端输出到sqlite数据库。krakenanalysis.py通过多数规则将kraken数据库中的分类标识分配给条形码组,其中条形码组根据其可分类读段中最常见的分类标记进行分类。使用脚本pure.py从参考对齐数据或系统发育标记计算条形码组纯度。计算机模拟血细胞计数:使用bowtie2v2.2.9对来自sic-reads数据库的读段进行对齐,其具有非常敏感和端到端的设置以引用所关注的序列(ar数据库获自gupta,s.k.等,《抗微生物剂与化疗(antimicrob.agentschemother.),58:212-220(2014),vf数据库获自毒力因子数据库下的核心vf基因(vfdb;chen,l.等,《核酸研究(nucleicacidsres.)》33:d325-328(2005),噬菌体序列数据库获自噬菌体基因组数据库,其在2016年5月在“http:”处之后“//www.ebi.ac.”之后“uk/genomes/phage.html”获取。然后对mapq>2过滤映射读段,以去除模糊映射读段。含有映射到数据库的读段的条形码组被注释为含有靶标序列,并且如果其在分类上被分类为纯度>0.8,则被输出用于进一步分析。为了产生转导潜力的热图,萃取与噬菌体和kraken分类的条形码组相关的所有读段并根据噬菌体类型分组。去除重复和接近重复的读段。热图强度计算如下:对于给定的一对细菌宿主,计数数据库中宿主-噬菌体-宿主连接的总数。为了按宿主丰度归一化数据,将此数除以与两个宿主关联的条形码组的总数。产生如图10a中所述的用于测序整个基因组的抗生素抗性网络:在旧金山海岸水微生物群落的sic-reads数据库中,使用最常与抗生素抗性相关的6个基因组的参考文献产生图10a中的抗生素抗性图。以下基因组(登记号)从ncbirefseq储存库下载:>cp003841.1|麦氏交替单胞菌(alteromonasmacleodii)atcc27126;>cp010434.1|枯草芽孢杆菌亚种spizizenii菌株nrs231;>cp000884.1|代尔夫特食酸菌(delftiaacidovorans)sph-1;>cp001918.1|阴沟肠杆菌(enterobactercloacae)亚种下水道atcc13047;>ae002098.2|脑膜炎奈瑟菌(neisseriameningitidis)mc58;和>ae017283.1|疮疱丙酸杆菌(propionibacteriumacnes)kpa171202。将这些基因组组合成单个fasta文件,并传递给短读段模拟器wgsimv0.3.2,所述模拟器产生各自具有0基本错误率的70bp读段的10m单端。使用bowtie2在“局部”模式下使用默认灵敏度设置将这些读段与抗生素抗性基因参考对齐。使用samtools(samtoolsview-b-f)去除所有未对齐的序列。将.sam格式的对齐序列导入cytoscapev3.4.0并且图10a所示的网络使用参考属和抗生素抗性基因分别作为网络靶标和来源而产生。图形边缘的暗度与数据中的连接总数成线性比例,其中较暗的线条具有更多的关联。产生用于图10b中的测序整个基因组的vf比率:图9b中的vf比率计算使用图中所示用于属的参考基因组再现。使用perl脚本ncbidownloader.pl从refseq数据库下载与这12个属相关的所有物种的完整基因组。将基因组合并到由属标记的fasta文件中。从这些参考文件中,python脚本(bargroupgenerator.py)产生每组200个读段的模拟条形码组,每个单端读段150bp长。为给定属产生的模拟条组的数量等于在旧金山海岸水样中为此属所识别的的条形码组的数量。然后使用具有默认灵敏度设置的“局部”对齐模式中的bowtie2v2.2.9将模拟条形码组读段与原始毒力因子数据库对齐。使用samtoolsv1.3.1(samtoolsview-b-f)去除未对齐的序列,并使用剩余的对齐读段产生图10b中所示的数据。结果:sic-seq工作流程:液滴微流体,其能够在每秒数千个单细胞上包封和进行生物反应,为单分子和单细胞应用提供了前所未有的潜力,包括均匀扩增、精确定量和深度测序单分子,并培养、筛选和测序单细胞的转录组。然而,单细胞基因组测序提出了独特的挑战,即每个细胞的基因组受到可能需要在酶处理之前将其去除的膜和蛋白质的保护。然而,通常用于基因组纯化的试剂,包括清洁剂、蛋白酶和高ph缓冲剂,但是可能对用于制备用于测序的酶有害,需要这些步骤分开进行。在sic-seq中,通过将细胞包覆在水凝胶微球(微凝胶)中来解决这个问题,所述水凝胶微球可以渗透水力直径小于孔径的分子,包括酶、清洁剂和小分子,但是在空间上捕获大分子如基因组。这允许在数百万个包覆的细胞上使用一系列“洗涤”,以进行细胞裂解和基因组处理的步骤,同时保持每个基因组的区室化。使用微凝胶和微流体处理步骤的组合,细胞被裂解,基因组被片段化,并且独特的条形码附接到所有片段,在几小时内处理>50,000个细胞的工作流程中。然后可以合并所有细胞的条形码化的片段并测序,并且通过条形码对读段进行分组,提供可以经受另外下游处理的单细胞基因组的库,包括人口统计表征和计算机模拟血细胞计数。图2中提供了sic-seq的工作流程图。有助于超高通量处理和单细胞测序的sic-seq的一个方面是标记源自相同基因组的dna片段,其具有所述细胞特有的序列标识符(条形码)。所得产物是嵌合的,包括与细胞基因组的随机片段共价连接的条形码序列。条形码允许通过共用序列识别属于给定细胞的所有读段。为了对许多单细胞的基因组片段进行独特地条形码化,使用了独特条形码序列的库。最近公开的条形码单细胞转录组的方法引入附接到固体珠粒或水凝胶球的条形码(klein,a.m.等,《细胞(cell)》,161:1187-1201(2015);macosko,ez等,《细胞》,161:1202-1214(2015),其中的每一个的公开以引用的方式并入本文中)。这些方法可以与本文描述的方法结合使用。然而,在本文示例的sic-seq的实施例中,含有条形码序列的液滴与要进行条形码化的基因组合并(参见,例如,lan,f.等,nat.commun.,7:11784(2016),其公开以引用的方式并入本文中)。为了制备条形码液滴库,使用微流体流动聚焦以0.1的速率将包括通过恒定序列侧接的15个随机碱的寡核苷酸包封(图1,图区a和图3a)(garstecki,p.等,《应用物理学报(appl.phys.lett.)》85:2649-2651(2004),其公开以引用的方式并入本文中)。然而,单个条形码分子通常不足以标记细胞的基因组,因此条形码分子通常在条形码过程之前被扩增。为实现此目的,使用pcr试剂和与条形码的恒定区互补的引物产生液滴,并且所述引物含有illuminap7流动细胞衔接子。然后对液滴进行热循环以通过数字液滴pcr扩增条形码序列。此方法以有效的方式在几小时内产生约1000万条形码液滴。在单细胞基因组可以被条形码化之前,其通常是从细胞体中物理分离和纯化的并且是片段化的。为了实现这一点,使用双流同向流动液滴制造器将单细胞包覆在琼脂糖微凝胶中,其将细胞悬浮液流与熔融琼脂糖流合并,形成由等体积的两种流组成的液滴(图3b和图1,图区a)。液滴制造器以约10khz运行,这允许在约20分钟内产生1000万个22μm的液滴,总体积的水性乳液分数约60μl。因此,液滴产生很快并且总体积消耗很小,允许以1:10的速率加载细胞以最小化多细胞包封;这降低了热循环期间聚结的可能性,混合了含有不同基因组的液滴,这将产生不希望的非单细胞条形码组。将熔融的琼脂糖液滴收集到冰上的pcr管中,使其固化。然后将固化微凝胶从油转移到水中,同时保持细胞的包封,然后将其进行裂解和基因组纯化。为了裂解细胞,将固化微凝胶在裂解酶的混合物中温育过夜,消化保护性微生物细胞壁(参见材料和方法)。然后将其在清洁剂和蛋白酶的混合物中温育30分钟,溶解脂质并消化蛋白质,仅保留高分子量基因组dna,这通过用sybr绿染料染色来验证。为了使基因组片段化并附着通用序列以充当pcr手柄,将固化微凝胶重新包封在反应中(图1,图区b)。重要的是,因为转座酶是二聚体,所以片段化的基因组作为大分子复合物保持完整,保持空间包覆在水凝胶网络内(图4)。在将基因组纯化并片段化后,将其条形码化以进行测序。使用将每个固化微凝胶包封在pcr试剂中的微流体装置,然后所述装置将固化微凝胶与条形码液滴合并(图3c和图1,图区c)。单分散微凝胶具有独特且有价值的特性,因为其是柔顺的,其可以通过微流体通道以高体积分数(>0.65)流动而不会堵塞,使其有序并定期流入液滴发生器。通过使液滴周期与微凝胶注射周期相匹配,可以实现液滴中微凝胶的有效加载,这种技术已被用于人类基因组单倍型分析和单细胞转录组测序,其具有最小的细胞损失,并且是商业化液滴微流体仪器的一部分。将含有片段化基因组和条形码化的液滴收集到pcr管中并进行热循环,通过由转座酶添加的pcr手柄通过互补性将条形码序列剪接到基因组片段上。此时,剪接片段含有illumina平台上用于测序所需的p5和p7illumina测序衔接子。在热循环期间,一些液滴聚结,产生对应于多个细胞的条形码聚类。在热循环结束时使用微量移液管去除这些聚结的液滴,并且然后将纯化的液滴化学破裂,并合并和制备其内容物用于测序(参见材料和方法)。测序后,按质量过滤读段并通过条形码分组,提供单细胞基因组序列数据。在人工微生物群落上验证sic-seq:sic-seq的目的是提供条形码组中捆绑的单细胞基因组序列;然后,此数据可用于微生物人口统计学表征,关联同一基因组内的所关注序列,以及作为基因组装配的潜在支架。要验证sic-seq产生单细胞的条形码组,其被施加到一种人工群落,其含有五种革兰氏阳性菌、三种革兰氏阴性菌、以及通过基因组dna含量在相同比例中混合的两种酵母(下表1)。为了确认裂解程序大体合理,这种混合物表示革兰氏阳性细菌和真菌,其通常难以裂解。使用sic-seq从此群落制备单细胞库,并将其在illuminamiseq上测序,在质量过滤后产生约600万个150bp的成对端读段。通过条形码对读段进行分组,并丢弃具有<50个读段的组,表示可能的pcr突变的条形码序列,并产生最终的48,989个条形码组(图5a)。每个条形码组理论上表示细胞的低覆盖度基因组,平均覆盖深度为1%,并且所有微生物的分布相似(图11)。表1:人工群落中十种细胞类型的列表生物体gc%克枯草芽孢杆菌43.8正新型隐球菌48.2n/a(酵母)粪肠球菌37.5正大肠杆菌56.8负发酵乳杆菌52.8正单核细胞增生性李斯特菌38.0正铜绿假单胞菌66.2负酿酒酵母38.4n/a(酵母)肠道沙门氏菌52.2负金黄色葡萄球菌32.7正为了确定条形码组是否确实对应于单细胞,将所有读段映射到三个已知物种的参考基因组。如果两个微生物位于相同的条形码组内,则读段将映射到两个基因组。组纯度得分定义为映射到最多映射参考的分数。组纯度得分的分布强烈偏移向高值,其中大多数纯度得分超过95%,表明大多数条形码组表示单细胞;即使考虑到十个物种的不同基因组大小(图5b和图12)以及在检查每个物种的纯度时(图13),此结果也是一致的。进一步检查具有低(<80%)纯度得分的罕见条形码组,并确定大多数这些条形码组表示其中两个细胞被包封成一个液滴以及偶尔聚结两个含有单细胞的液滴(图14)的罕见情况。为了确定sic-seq条形码丰度是否反映数据集中的生物体丰度,比较通过短读段对齐、宏基因组序列分类和亮场显微术下的计数计算的丰度估计值(图5c和图15)。发现当合并读段并使用这些方法大量分析时,并且还当基于组中最常映射的物种将物种标识分配给每个条形码时,所有方法都是合理的一致。这表明sic-seq能够估计微生物群体中的物种丰度,这与公认的宏基因组方法一致。为了研究sic-seq中的覆盖分布偏差,针对每种微生物从所有条形码组聚集的读段绘制归一化覆盖分布(图5d,图5e和图16)。除了由于标准微生物群落内细胞的基因组dna丰度之间的差异导致的覆盖度缺口之外,未观察到显著的覆盖度偏差。这表明条形码组内每个基因组的取样是随机的,因此当所有组重叠时,获得均匀分布。进一步检查各个条形码组中的读段分布并且没有发现显著的偏差(图6a,图6b)。此外,偏差是最小的,因为每个基因组以约65μl的微小体积扩增,其已经显示减少诱导指数扩增失控的偏差。此外,测序库由约50,000个扩增的基因组构成,因此,每个基因组的扩增可以受到微小体积的限制,同时仍然产生足够的总dna用于测序。sic-seq产生可以使用计算机模拟血细胞计数分析的新型数据:使用sic-seq产生的基因组序列根据单细胞分组,其与短读段宏基因组测序的序列互补。现有的计算工具不适合分析这些数据,因为其没有使用sic-seq独有的单细胞条形码信息。为了解决这个问题,使用了一种新的序列分析流程,其中读段被分层组织为条形码组,产生单细胞读段据库(sic-reads)(图7)。为了构建sic-reads,按质量过滤原始序列,按条形码分组,并使用系统发育分析仪估计每组的分类学分类。还基于分析器可分类的读段估计纯度得分,指定等于在可分类集合内映射到显性分类的读段的分数的值。条形码组和读段的其它特性,如对应于抗生素抗性基因的序列的存在,可以在分析期间发现时添加到数据库中。sic-reads中存在的单细胞基因组的集合为在单细胞内发现序列之间的关联提供了新的机会,这一过程称为计算机模拟血细胞计数。sic-reads包括单细胞基因组的多维集合,其可以在计算机中进行分选,类似于单细胞上流式细胞术的常规方法。虽然流式细胞术要求先验地选择靶标生物标志物并且可以以使用的生物标志物的数量受到限制,但是计算机模拟血细胞计数可以进行多次并且具有所需的尽可能多的序列生物标志物。可以重复对数据库进行分选以挖掘不同基因序列和结构之间的相关性。此外,随着新关联的学习,可以定义新的分选参数,无需重复实验即可实现新的发现,最终仅受单细胞数据库的完整性和准确性的限制。为了说明sic-seq和计算机模拟血细胞计数的功效,对从旧金山的沿海海水中回收的微生物群落进行了测序(参见材料和方法)。在质量过滤后获得约800万个长度为150bp的读段(表示约55%的原始读段,其中产生sic-reads数据库(图7)。将601,348(6.89%)个读段分类为表示99.8%细菌、0.04%古细菌和0.16%病毒的分类群(图8,图区a)。条形码组根据其含有的读段被分配了一个分类学分类,遵循超过10%的读段必须具有分类的规则,并且所述组根据具有最多负载读段的分类单元进行分类。根据对照样品(约94%),大多数条形码组基于可分类的序列(约91%)估计为高纯度(图8,图区b)。使用此数据,通过探索微生物群落中抗生素抗性、毒力因子和噬菌体序列的分布来证明计算机模拟血细胞计数。栖居于旧金山海岸线的微生物中抗生素抗性的分类学分布:抗生素抗性(ar)已经变得越来越普遍并且对全球人类健康构成重大威胁。因为抗生素是对抗细菌感染的主要工具,了解ar基因如何在自然环境中传播对于维持细菌疾病的有效对策至关重要。微生物可通过多种机制获得ar,包括突变、抗性基因的采集,甚至基因缺失。虽然可以通过短读测序在大多数环境中识别ar基因,但是关于其如何在分类群中分布的信息很少,因为获得这些信息通常需要对单一物种进行测试或全基因组测序;然而,大多数物种都是不可开发的,排除此类分析。sic-seq提供了一个独特的机会来表征样品中所有物种中ar基因的分布,包括不可复制的物种,因为物种可以根据条形码组中的读段进行分类,并且然后与也存在的ar基因相关联。为了确定数据集中分类群中ar基因的分布,在sic-reads数据库中搜索已知的ar基因,发现1,081个(0.012%的读段),表示108个(0.30%)条形码组。ar基因的分类学分布具有清晰的结构(图9a和图10,图区a);与分离和测序菌株的环境相比,在天然海岸线环境中预期存在差异。与ar相关的最丰富的分类群并不是整个群落中最丰富的分类群,这表明在此群落中某些分类群往往更多地与ar基因相关联。例如,氨基糖苷抗性主要见于交替单胞菌属物种(alteromonasspp.),而β内酰胺类抗性广泛传播,在5个分类群中的4个中都发现。虽然不打算受任何特定理论的束缚,但β-内酰胺的广谱活性有可能促使人们大量使用,并相应地导致旧金山沿海微生物的广泛抗性。另一方面,氨基糖苷抗生素不常用于人类,并且因此对其的抗性很少,识别的实例可能表示天然发现于交替单胞菌属中的基因,其主要目的是不避免氨基糖苷抗生素介导的杀伤。将毒力因子与微生物群落中的宿主细菌联系起来:毒力因子(vf),如ar基因,对于确定特定微生物对人类健康的威胁非常重要。许多机会性病原体存在于环境中的自然群落中,并且当传播给合适的宿主时会引起爆发。因此,监测和检测潜在的致病微生物对公众健康很重要。虽然宏基因组学鸟枪法测序或dna微阵列方法可以检测到群落中vf的丰度,但其无法确定哪些微生物携带它们,或者同一个微生物中是否存在多个vf-这两者都是物种致病潜力的重要决定因素。同样,sic-seq提供了一个独特的机会来表征群落中的vf并将其与特定宿主物种联系起来。在沿海微生物群落数据库中搜索已知的vf基因,并在101个(0.28%)条形码组中的1,949个(0.022%)读段中产生匹配,所述条形码组由分布在13个微生物属中的29个普遍的vf组成。具有vf的分类群的丰度并未反映总群体的数量,这表明某些属的vf比其它属更多。为了量化这一点,计算vf比率,含有vf的条形码组的数量与所述物种的群落中的条形码总数之间的比率,并且将结果归一化为最高vf比率用于比较(图9b)。嗜血杆菌属和埃希氏杆菌属在所有物种中都很突出,这两种物种都是已知的机会性人类病原体。仔细观察后,在嗜血杆菌属中检测到的主要vf是脂寡糖,其是嗜血杆菌属外膜的主要成分,也是宿主免疫逃避的重要决定因素。在埃希氏杆菌属中,检测到的主要vf是k1胶囊和iii型分泌系统,两者通常存在于毒性菌株中。将旧金山海岸线群落的vf比率与公开可用的全基因组的vf比率进行比较,并进行下采样以匹配每个细胞的读段深度(图10,图区b),发现用于公开基因组的比率较高,表明在当前测序的基因组中偏差朝向致病菌株。确定微生物群落中细菌之间的转导潜力:许多毒性细菌菌株被认为是由噬菌体交叉感染辅助的水平基因转移引起的。噬菌体可以修饰其宿主的基因组,留下其自身基因组后的拷贝,或者在一个被认为是产生毒性细菌菌株的重要机制的过程中将一个物种的片段运输到另一个物种,称为转导。然而,与ar和vf基因一样,表征这些移动元件的分布在生态环境中具有挑战性,因为在特定宿主内对外源基因组片段的可靠识别需要对单物种或单细胞的培养物进行测序。尽管如此,此信息对于理解细菌如何一般地转移基因材料以及特别是通过这种机制可能出现的毒性新菌株非常有价值。为了探索微生物群落中的转导,在旧金山沿海群落的sic-reads数据库中搜索含有噬菌体序列的条形码组。在细菌基因组中发现的噬菌体序列证明其可能潜在地感染宿主,这种关联通常对于难以培养的微生物和其可能无法培养的感染噬菌体非常困难。在6,805(0.078%)个读段中发现匹配表示260个(0.72%)条形码组和106个噬菌体基因组。由于转导可以在同一噬菌体可感染的两种宿主细胞之间发生,转导的可能性取决于有多少种类型的噬菌体感染两种宿主,以及这些噬菌体感染两种分类群的程度。为了使其可视化,绘制了检测到与两个细菌分类群中的相同噬菌体匹配的序列的次数的总和,通过这些属中的条形码组的数量归一化(图9c)。根据此分析,在此分析中与分类群最密切相关的代尔夫特菌和奈瑟菌属具有最高的转导潜力。然而,sic-seq检测这些序列并将其与单个基因组相关联的能力为研究噬菌体-宿主相互作用提供了一种新颖而有效的方法。这种相互作用未被充分研究,因为缺乏用于测序单细胞的高通量方法,但这对于理解微生物群落动态是至关重要的。实例2:通过malbac反应的单细胞的微流体条形码在此实例中,使用具有已知组成的合成细胞的群落来评估扩增和条形码性能。包封在水凝胶中的细胞可用于代替本方法中描述的细胞悬浮液。材料和方法:微流体装置:为了制造微流体装置,将聚(二甲基硅氧烷)(道康宁,sylgard184)倾倒在使用uv光刻在硅晶片(universitywafer)上图案化的负光致抗蚀剂(microchem,目录号su-83025)上。将pdms装置在烘箱中固化1小时,用金属刮刀萃取,并用0.75mm活检芯(世界精确度仪器,目录号504529)打孔以产生入口和出口。使用氧等离子体清洁剂(harrickplasma)和用aquapel(ppgindustries)处理的通道将装置粘合到玻璃载片上,并在80℃下烘烤10分钟以使其疏水。条形码化乳液:通过非对称数字pcr方法制备条形码化乳液,其中条形码化寡核苷酸在含有pcr试剂的液滴中作为单分子扩增。非对称pcr有利于单链条形码化产物的生产。将在0.01pm浓度下的条形码化寡核苷酸bd23(gtatgcgacttcagtacatcgtcccgatgctctgcacagtcgaccgctgannnnnnnnnnnnnnncagcgatcccgagtcagatcgatgccactgagacctgtgagtgatggttgaggtagtgtggag)(seqidno:9)(idt)添加到含有以下的pcr反应混合物中:1xneb福讯热启动挠曲主混合物(neb,目录号m0536l)、2%(w/v)tween20(西格玛-阿尔德里奇,目录号p9416)、5%(w/v)peg-6000(圣克鲁兹生物技术,目录号sc-302016)、0.2μm限制性引物bd27(ctccacactacctcaaccatcactcacaggtctcagtggc)(seqidno:10)和1μm过量引物bd28(gtatgcgacttcagtacatcgtcccgatgctctgcaca)(seqidno:11)(idt)。将具有2%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂(008-氟表面活性剂,ran技术)的pcr混合物和hfe-7500氟化油(3m)装入单独的1ml注射器(bd)中并分别以300和500μl/hr注入使用由定制python脚本(“https:”之后“//github.”之后“com/abatelab/泵-控制-程序(pump-control-program)”)控制的注射泵(newera,目录号ne-501)的同向流动液滴制造器(图21)。将乳液收集在pcr管中,并通过移液管去除乳液下面的油,并用具有5%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂的fc-40氟化油(西格玛-阿尔德里奇,目录号51142-49-5)代替,以改善热稳定性。使用以下程序对乳液进行热循环(拜耳雷德,t100):98℃3分钟,之后进行40个循环,其中每秒2℃的升温速率为98℃10秒、63℃20秒,以及72℃20秒,之后在12℃下保持。使用hfe-7500油中的10xsybrgreeni(赛默飞世尔科技)进行的荧光dna染色用于在荧光显微镜(生命技术,目录号amafd1000)下定量条形码化包封率。细胞悬浮液的制备:为了通过sic-seq工作流程制备用于加工的人工群落,将冷冻的细胞原料(兹莫研究,目录号d6300)在室温水浴中温和解冻。通过在显微镜下手动细胞计数测定细胞浓度,并稀释至适当浓度以进行单细胞包封。将计算的细胞溶液体积转移至1.5ml离心管(飞世尔科技公司)中并在1mlpbs中洗涤两次。将细胞再悬于1mmtris-hclph8.0(teknova)的1ml溶液中。单细胞的malbac扩增:制备含有以下的malbac扩增混合物:2xthemopol缓冲液(neb,目录号b9004s)、2%(v/v)tween20、6%(w/v)peg-6000、1.2mmdntps(neb,目录号n0447s)、0.64μmgat-8n引物(gtgagtgatggttgaggtagtgtggagnnnnnnnn)(seqidno:1)(idt)、4mmmgso4(neb,目录号b1003s)和0.12u/μl深通气孔(外-)聚合酶(neb,目录号m0259s)。将具有2%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂的malbac扩增混合物、细胞悬浮液和hfe-7500氟化油(3m)装入单独的1ml注射器中并分别以200、200、1000μl/hr注入同向流动液滴制造器(图21)。将乳液收集在pcr管中,并通过移液管去除乳液下面的油,并用具有5%(w/w)peg-pfpe两亲嵌段共聚物表面活性剂的fc-40氟化油代替,以改善热稳定性。使用以下程序对乳液进行热循环(拜耳雷德(bio-rad),t100):95℃5分钟,之后进行8个循环,其20℃50秒、30℃50秒、40℃45秒、50℃45秒、65℃4分钟、95℃20秒,并且58℃20秒,之后在12℃下保持。尽管此具体实例没有使用水凝胶,其指出的是水凝胶可以被整合进细胞悬浮液的地方的工作流程。扩增细胞的微流体条形码:将malbac-扩增细胞液滴、条形码化液滴和300μl延伸溶液,其含义1xthemopol缓冲液、1%(v/v)tween20、3%(w/v)peg-6000、0.5mmdntps、2mmmgso4和0.06u/μl深通气孔(外-)聚合酶各自装入1ml注射器中并注入合并装置,如图22中所示。使用具有2%(w/w)008-含氟表面活性剂的hfe-7500氟化油作为乳液的连续相。使用连接到冷阴极荧光逆变器和dc电源(mastech)的电极实现条形码和细胞液滴乳液的合并。电源下的2.0v电压在电极处产生约2kv的ac电势,这致使触摸液滴合并。将乳液收集在pcr管中,并在单循环pcr之前用5%(w/w)008-含氟表面活性剂替换为具有fc-40的hfe-7500,其具有以下方案:95℃2分钟,55℃30秒,72℃5分钟,并且然后保持12℃。热循环后,通过添加20μl全氟辛醇并在微型离心机中短暂离心来破坏乳液。收集上层水相,并纯化dna库,并使用agencourtampurexp珠粒(贝克曼库尔特公司)以0.5x体积比的珠粒与pcr产物去除引物。将大珠粒结合的dna片段在水中洗脱。单链发夹环的消化:无条形码的单链malbac扩增子通过绿豆核酸酶、核酸内切酶选择性靶标ssdna来消化。制备消化反应,其含有条形码化的pcr产物、1x绿豆核酸酶反应缓冲液(neb,目录号m0250s)和0.033u/μl绿豆核酸酶(neb,目录号m0250s)。将反应在30℃温育30分钟,并且然后通过添加0.5%(w/v)十二烷基硫酸钠(西格玛-阿尔德里奇)终止到0.02%(w/v)的最终浓度。使用agencourtampurexp珠粒以0.5x体积比的珠粒与pcr产物进行大小选择以去除小的消化产物。条形码化的基因组dna的富集pcr:通过pcr在溶液中扩增根据消化反应的条形码化的dna产物,所述溶液含有1xinvitrogen铂多重pcr主混合物(赛默飞世尔科技,目录号4464268)、0.5μmgat-com引物(gtgagtgatggttgaggtagtgtggag)(seqidno:2)(idt)和0.5μmbd24引物(gtatgcgacttcagtacatcgtcccgatgctctgcacagtcgaccgctga)(seqidno:12)(idt),其使用以下方案:95℃1分钟,25个循环95℃20秒,80℃5秒,55℃20秒(0.3℃/s升温速率)、72℃3分钟,之后在72℃下最后5分钟延伸,其中12℃保持。使用兹莫dna清洁&浓缩器-5试剂盒(zymodnaclean&concentrator-5kit)(兹莫研究)纯化pcr产物。使用agencourtampurexp珠粒以0.5x体积比的珠粒与pcr产物进行片段大小选择以去除pcr引物。用生物分析仪2100仪器和高灵敏度dna芯片(安捷伦)定量之前(图23)和之后(图24)大小选择的dna片段。条形码化的dsdna片段具有约1500bp的平均长度。库制备和下一代测序:根据制造商方案,nexteraxtdna库制备试剂盒(illumina,目录号fc-131-1024)用于制备测序库。简言之,将600pg条形码化的dna用作标记反应的输入。中和后,使用以下通过12个pcr循环扩增转座子:nexterapcr主混合物、0.2μm定制引物bd29(aatgatacggcgaccaccgagatctacacgtatgcgacttcagtacatcgtcccgatgctctgcacagtcgaccgctga(seqidno:13),含有p5测序衔接子)(idt)和0.2μmilluminanexteran706引物(caagcagaagacggcatacgagatcatgcctagtctcgtgggctcgg(seqidno:14),含有p7测序衔接子)(idt)。使用bluepippin仪器(sagescience))进行300-600bpdna片段的最终大小选择。通过量子位(qubit)3.0荧光计(invitrogen)和具有高灵敏度dna芯片的生物分析仪2100仪器对库进行定量。使用300循环miseq试剂nano试剂盒v2(illumina,目录号ms-103-1001)在miseq仪器上对库进行测序。根据制造商方案使用定制读段1(cagcgatcccgagtcagatcgatgccactgagacctgtgagtgatggttgaggtagtgtggag)(seqidno:15)(idt)和指数1(gtatgcgacttcagtacatcgtcccgatgctctgcacagtcgaccgctga)(seqidno:12)(idt)引物。产生sic读段数据库:miseq产生的fastq文件的原始读段按质量过滤,并使用python脚本barcodecleanup.py按条形码序列分组。如果超过20%的碱基的q-得分低于q20,则丢弃给定的读段,并丢弃与含有少于50个读段的条形码相关的所有读段。此步骤确保表示单细胞的所有条形码组含有足够数量的高质量读段。将得到的读段输出到sqlite数据库中的表中,其含有条形码序列、条形码组大小、唯一读段id号和读段序列的领域。对于合成细胞群体实验,使用具有默认设置的bowtie2v2.2.9对齐读段,并且使用每次读段的相关对齐信息更新sqlite表。结果:库测序产生800,189个读段,其中596,357个(74.5%)属于含有最少50个读段的条形码组。获得2,186个条形码组(最少50个读段)。属于这些条形码组的读段中,99.3%与来自合成群落的十个参考基因组中的一个对齐。用于评估条形码之间的交叉污染的纯度度量定义如下:条形码组的纯度定义为条形码组中对齐的读段的分数,其与所述组中最常见的参考基因组对齐。纯度为1.0意指给定条形码组中的所有读段都与同一基因组对齐。图25显示了实验中所有条形码组的纯度得分的分布(每组最少50个读段)。这些组通常是高纯度的,其中平均纯度为0.950。具有更多读段的条形码保持高纯度(图26)。各个条形码组内的读段在整个参考基因组中对齐(图27),这是malbac的低偏差扩增特征的结果。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1