一种树状大分子功能化聚合物的制备方法及其应用与流程
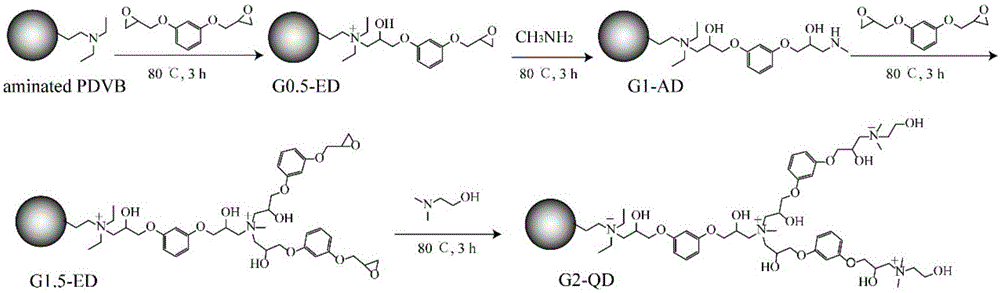
本发明涉及一种对酸性非甾体抗炎药具有选择性的新型树状大分子功能化聚合物的制备和应用,属于色谱新材料和样品前处理领域。
背景技术:
非甾体抗炎药(non-steroidanti-inflammatorydrugs,nsaids)是一类具有抗炎、止痛和退热作用的药物,被人类和牲畜广泛应用。有证据表明该类药物具有很大的肾毒性,例如南亚的秃鹫由于食用了残留有双氯芬酸的牛尸体而大量死于肾衰竭(achemtrustreport(2014))。非甾体抗炎药也能够增加心血管不良事件和并发症(pharmaceuticals3(2010)2146-2162)。由于该类药物的使用量大,副作用大,因此对复杂样品中非甾体抗炎药的准确快速检测显得尤为重要。
由于药物在生物和环境样品中的含量很低(ngml-1~μgml-1),同时含有很多干扰物质,因此样品的富集和净化是样品制备的关键步骤。由于固相萃取技术具有吸附剂材料多样性、回收率好、有机溶剂用量少等优点,是最常用的样品前处理技术。传统的固相萃取材料通常只有一种色谱保留模式–反相作用(如c18,st-dvb)或者离子交换作用(离子交换树脂),选择性差。混合模式吸附剂集反相作用和离子交换作用于一体,改善了对目标分析物的选择性和保留能力,可以更有效的去除杂质,获得高灵敏度和低背景干扰。
增加混合模式吸附剂材料的作用位点可以改善吸附剂材料的选择性和萃取效率。树状大分子是一种高度支化、呈辐射状的新型功能高分子,内部具有纳米空腔和丰富的活性位点,外围具有可以修饰的官能团,在纳米催化、药物运输、化学传感器、污水处理、膜材料等方面有潜在应用。将树状大分子应用于固相萃取技术可以显著提高材料的作用位点。目前的文献报道中,还未有以间苯二酚二缩水甘油醚和甲胺为功能单体,通过逐步接枝的方法,在聚合物表面修饰树状大分子作为反相/阴离子交换混合模式吸附剂富集和净化复杂样品中的酸性非甾体抗炎药的报道。
技术实现要素:
本发明的目的是提供一种对酸性非甾体抗炎药具有超高选择性的树状大分子功能化聚合物的制备方法。
一种树状大分子功能化聚合物的制备方法,所述方法包括以下步骤:
步骤一、采用pickering乳液聚合法制备pdvb-sio2(纳米二氧化硅稳定的聚二乙烯基苯)复合微球;
步骤二、将所得pdvb-sio2复合微球用氢氟酸浸泡去除纳米sio2,分离得到pdvb微球;
步骤三、采用逐步反应在聚合物表面接枝树状大分子
(1)将pdvb微球分散在甲醇中,pdvb的浓度为0.05~0.1g/ml;加入间苯二酚二缩水甘油醚,于60~80℃反应3~5h,洗涤、真空干燥,得到末端官能团为环氧的树状大分子修饰的聚合物(g0.5-ed);
(2)将所得g0.5-ed分散在乙醇中,g0.5-ed的浓度为0.05~0.1g/ml;加入甲胺,于60~80℃反应3~5h,洗涤、真空干燥,得到末端官能团为胺基的树状大分子修饰的聚合物(g1-ad);
(3)重复所述(1)和(2),得到不同代数的树状大分子功能化聚合物;
步骤四、环氧端基的季氨化反应
将所得末端官能团为环氧的树状大分子功能化聚合物gn-ed(n=m-0.5,m为≥1的整数)分散在乙醇中,gn-ed浓度为0.05~0.1g/ml;加入叔胺化合物(优选为n,n-二甲基乙醇胺),于60~80℃反应3~5h,得到端基为季铵基团的树状大分子功能化聚合物gm-qd(m为≥1的整数)。
所述pickering乳液聚合法以纳米sio2为乳化剂,以甲基丙烯酸二乙氨基乙酯(deaema)为单体;以二乙烯基苯为交联剂;以偶氮二异丁腈为引发剂;以甲苯和1-十二醇为致孔剂,在50~70℃聚合反应12-36h;所述1-十二醇与甲苯的摩尔比为1:4~5;纳米sio2与油相(油相为甲基丙烯酸二乙胺基乙酯、二乙烯基苯、甲苯、1-十二醇和偶氮二异丁腈混合溶液)质量比为1%~4%;
所述单体、交联剂、致孔剂、引发剂的摩尔比为1:1~5:2~8:0.02~0.1。
步骤二中,所述pdvb微球的粒径优选为15~35μm。
步骤二中,优选还包括将所得pdvb微球进行以下抽提的步骤:采用甲醇、乙腈、乙醇、丙酮中的一种或一种以上的溶液为提取溶剂,索氏抽提,抽提温度为80~120℃,除去聚合物中未反应的物质和低聚物;
步骤二中,优选所述氢氟酸的质量浓度为20%~40%,不加入或其中可加入甲醇或丙酮溶剂中的一种或二种。
步骤三(1)中,优选所述间苯二酚二缩水甘油醚在总反应物中的体积溶度为5%~15%;
步骤三(2)中,优选所述甲胺在总反应物中的体积浓度为5%~20%;
步骤四中,优选叔胺化合物在总反应物中的浓度为5~15%。
本发明还提供上述方法制备得到的树状大分子功能化聚合物。
本发明还提供上述树状大分子功能化聚合物作为反相/阴离子交换混合模式吸附剂材料在选择性富集净化复杂样品中的痕量酸性有机物中的应用;所述酸性有机物包括酸性非甾体抗炎药。
所述树状大分子功能化聚合物作为固相萃取柱的填料。
优选地,所述样品包括液体样品和固体样品的提取溶液;所述液体样品包括饮用水、牛奶、河水、污水、血液、尿液;所述固体样品包括底泥、土壤、肉类。
本发明所述树状大分子修饰的聚合物对水杨酸、布洛芬、萘普生、酮洛酚、双氯芬酸等酸性非甾体抗炎药具有很高的选择性,能够从溶液中吸附该类药物,通过选择合适的淋洗条件除去吸附的干扰物质而保留目标物。将其用作样品预处理材料,可以使目标化合物得到高回收率和超高的净化效果。
由于采用树状大分子修饰,极大的增加了吸附剂的离子交换容量,吸附能力增强。由于树状大分子修饰的聚合物作为吸附剂具有反相和离子交换双重保留机理,通过选择合适的淋洗条件,可以去除干扰物质而只保留目标分析物,克服了传统聚合物回收率低、净化效果差的问题。
本发明的优点是:提供了一种新型高容量树状大分子功能化聚合物的制备方法及应用。采用逐步接枝方法在聚合物表面修饰不同代数的树状大分子,充分利用了树状大分子的高度分支结构、高密度活性位点来增加材料的吸附位点,提高吸附效率和选择性。将树状大分子功能化聚合物作为固相萃取柱填料,可以分离纯化复杂基质中的酸性非甾体抗炎药,能够有效减小基质干扰,能够快速准确地对复杂样品进行检测。
附图说明:
图1是聚合物的合成示意图;
图2是碱性(卡马西平和阿米替林)、中性(氢化可的松)和酸性(布洛芬)药物的洗脱曲线。
具体实施方式
①将单体、交联剂和引发剂溶解在致孔剂中,制备成溶液a,向溶液a中通氮气;,优选15min,除去溶液a中的氧分子;单体、交联剂、致孔剂、引发剂的摩尔比为1:1~5:2~8:0.02~0.1;单体优选为甲基丙烯酸二乙氨基乙酯(deaema);交联剂优选为二乙烯基苯;引发剂优选为偶氮二异丁腈;致孔剂优选为甲苯和1-十二醇,其中1-十二醇与甲苯的摩尔比为1:4~5;sio2粒径小于50nm。
②将纳米sio2分散到去离子水中,称为分散液b,纳米sio2质量浓度为3~15mgml-1(优选浓度为12mg/ml)。然后向10ml分散液b中加入3~5ml溶液a,6000rpm匀浆分散1min。通氮气2min排出体系中的氧分子后密封。将得到的pickering乳液在50~70℃聚合反应12~36h。
③反应结束后,采用离心分离或抽滤,得到白色聚合物和sio2复合物材料(pdvb-sio2)。
④所得到的pdvb-sio2复合材料用氢氟酸浸泡12~24h,分离得到白色聚合物微球(pdvb)。
⑤筛分、沉降,得到粒径为15~35μm的聚合物微球。
⑥所得到的材料采用甲醇、乙腈、乙醇、丙酮中的一种或一种以上的混合液为提取溶剂进行索氏抽提,抽提温度为80~120℃,除去聚合物中未反应的物质和低聚物。
⑦采用逐步反应修饰树状大分子
将pdvb分散在90%甲醇中,pdvb的浓度为0.05~0.1g/ml。加入间苯二酚二缩水甘油醚于60~80℃反应3~5h,得到末端官能团为环氧的树状大分子修饰的聚合物(g0.5-ed),用甲醇和丙酮洗涤g0.5-ed,并于真空干燥。
然后将g0.5-ed分散在乙醇中,g0.5-ed的浓度为0.05~0.1g/ml。加入甲胺溶液于60~80℃反应3~5h,得到末端官能团为胺基的树状大分子修饰的聚合物(g1-ad),用甲醇和丙酮洗涤g1-ad,并于真空干燥。重复上述两步反应,得到不同代数树状大分子功能化的聚合物;
⑧环氧端基的季氨化反应
将制备得到的末端为环氧的树状大分子功能化聚合物gn-ed(n=m-0.5,m为≥1的整数)分散在70%乙醇溶液中,gn-ed浓度为0.05~0.1g/ml。加入n,n-二甲基乙醇胺于60~80℃反应3~5h,得到端基为季铵基团的高容量树状大分子功能化聚合物gm-qd(m为≥1的整数)。
其中,制备树状大分子功能化聚合物的最佳条件为:在上述步骤①中功能单体为甲基丙烯酸二乙氨基乙酯,交联剂为二乙烯基苯,引发剂为偶氮二异丁腈,单体:交联剂:致孔剂:引发剂摩尔比为1:4:6.5:0.07,致孔剂中1-十二醇和甲苯的摩尔比为1:4.7。
实施例1
将10mmol(2.0ml)甲基丙烯酸二乙氨基乙基酯、40mmol(5.66ml)二乙烯基苯和115mg偶氮二异丁腈溶解到含有甲苯(5.68ml)和1-十二醇(2.12g)的致孔剂溶液中,制备成溶液a,然后向溶液a中通氮气15min除去氧分子。将120mg粒径12nm的sio2分散到10ml水中,超声10min使纳米sio2分散,然后加入4ml溶液a,采用匀浆机6000rpm搅拌1min,然后通氮气2min除去体系上方的空气后密封,将得到的pickering乳液在60℃聚合反应24h。反应结束后抽滤,然后过400目不锈钢筛,再用丙酮沉降两次,每次10min,得到粒径为15~35μm的聚合物pdvb-sio2。然后取2gpdvb-sio2用10ml40%的氢氟酸浸泡24h,除去表面的sio2得到pdvb微球。抽滤,并用去离子水洗涤至中性。以甲醇为提取溶剂,索氏提取24h,然后于真空干燥箱中60℃干燥12h。
将2.0gpdvb分散在20ml90%甲醇中,加入2.5ml间苯二酚二缩水甘油醚于80℃反应3h,得到末端官能团为环氧的树状大分子修饰的聚合物(g0.5-ed),用甲醇和丙酮洗涤g0.5-ed,并真空干燥。然后将g0.5-ed(2.0g)分散在20ml乙醇中,加入5ml甲胺溶液于80℃反应3h,得到末端官能团为胺基的树状大分子修饰的聚合物(g1-ad),用甲醇和丙酮洗涤g1-ad,并真空干燥。重复上述两步反应,得到不同代数树状大分子功能化的聚合物;
将制备得到的末端为环氧的树状大分子功能化聚合物2.0ggn-ed(n=m-0.5,m为≥1整数)分散在20ml70%乙醇溶液中,加入2.5mln,n-二甲基乙醇胺于80℃反应3h,得到端基为季铵基团的树状大分子功能化聚合物gm-qd,(m为≥1整数)。不同代数的树状大分子功能化聚合物的比表面积和孔体积结果见表1。
实施例2
将50mgpdvb装入3-mlspe柱管,先用5ml甲醇和5ml去离子水平衡,将7mg布洛芬用30ml10mm的磷酸盐缓冲液(ph7.0)溶解并上样。抽干30min,用5ml甲醇淋洗,然后用5ml1%甲酸的甲醇溶液洗脱布洛芬,收集洗脱溶液,用甲醇/水(1:1)稀释,定容到250ml,用高效液相色谱测定,计算离子交换容量。g1-qd、g2-qd、g3-qd、g4-qd和g5-qd的离子交换容量测定方法与pdvb的相同。不同代数的树状大分子功能化聚合物的离子交换容量结果见表1。
表1不同代数的树状大分子功能化聚合物的比表面积、孔体积及离子交换容量测定结果
通过表1,可以看出随着树状大分子在聚合物孔道中生长,(1)聚合物的比表面积和孔体积逐渐减小,(2)材料的离子交换容量逐渐增加,直至第四代树状大分子修饰的聚合物(g4-qd)的离子交换容量最大,而g5-qd的离子交换容量与g4-qd相比有所下降。所以选择g4-qd作为固相萃取柱的填料。
实施例3
将200mgg4-qd装入3-mlspe柱管,先用5ml甲醇和5ml10mm磷酸盐缓冲溶液平衡,将10ml5μgml-1的卡马西平(caz)、阿米替林(ami)、氢化可的松(hyc)和布洛芬(ibp)的磷酸盐缓冲液(ph7.0)以1mlmin-1的速度上样。抽干30min,先用5ml甲醇淋洗,然后用5ml1%甲酸的甲醇溶液洗脱,收集淋洗溶液和洗脱液,每毫升作为一段溜份,氮气吹干,定容至0.5ml,用hplc-uv分析。不同物质的流出曲线见图2,通过图2看出可以用2ml甲醇就可以将卡马西平(caz)、阿米替林(amt)和氢化可的松(hyc)从固相萃取柱上洗脱下来,而必须使用含有甲酸的甲醇溶液才可以将布洛芬洗脱,结果说明通过选择合适的条件,树状大分子功能化聚合物可以选择性保留酸性布洛芬。
- 还没有人留言评论。精彩留言会获得点赞!