一种支化程度可控的聚烯烃材料的合成方法与流程
本发明涉及聚烯烃领域技术,具体涉及一种支化程度可控的聚烯烃材料的合成方法。
背景技术:
聚烯烃是开发最早、产量最高、应用最广的高分子材料。聚烯烃又是一类成分最为简单的高分子材料,从组成成分上来看,聚烯烃仅仅由碳元素和氢元素两种元素组成。尽管组成成分简单,聚烯烃的材料却可以因其化学结构的变化而呈现千变万化的性能。较常见的聚烯烃材料包括聚乙烯、聚丙烯、聚丁二烯、聚α-烯烃等各种类型,在生活生产的各个领域扮演着无可替代的重要角色。从高分子物理化学角度上看,聚烯烃材料的特性主要受其化学结构、分子量及其分布、填料、加工方式等影响。在诸多影响因素之中,具有决定性作用的是聚烯烃材料的化学结构。尽管这一类材料通常只由碳氢两种元素组成,其聚合物的链结构却千变万化。各类烯烃材料具有不同的支链结构,而这些支链的长度、密度、排布方式等会对材料性能产生深远的影响。以聚乙烯材料为例,依支化程度及分子量不同,可分为超高分子量聚乙烯(uhmwpe)、高密度聚乙烯(hdpe)、低密度线型聚乙烯(lldpe)、低密度聚乙烯(ldpe)等不同材料。一般情况下,聚烯烃材料的支化程度越高,材料的结晶度也就越低,对应的粘度越低、流动性也就更好,具有更好的可加工性能,也可用于涂层、润滑剂、交联剂等领域。
受目前合成方法的限制,很难合成具有较高支化程度的聚烯烃。尽管guanzhibin等人在90年代就提出了采用“链行走聚合”方法合成具有较高支化程度的聚烯烃材料(science,vol283,1999),这一类方法受所用反应机理及催化剂的限制,很难控制聚合物的支化结构,所得聚烯烃材料的支化长度受统计学规律制约而呈现随机分布,从而影响这一类材料的性能。因而,设计新的合成方法以合成具有高度可控、高度精确支化结构的聚烯烃材料是这一领域发展的重中之重。
技术实现要素:
本发明要解决的技术问题是:针对现有技术不足,提供一种支化程度可控的聚烯烃材料的合成方法,具体为一种精确可控的超支化聚烯烃的合成方法,通过控制反应过程中的各参数,可是实现对聚烯烃支化程度、分子量及其分布的控制,从而实现其他方法无法达到的具有精确可控支化结构的超支化聚烯烃的合成。
本发明的目的是通过以下技术方案实现的:
本发明提供了一种支化程度可控的聚烯烃材料的合成方法,包括以下步骤:
将环状烯烃单体在催化剂条件下进行聚合反应;
所述环状烯烃单体如式i所示:
优选地,所述聚合反应中,环状烯烃单体的摩尔浓度为0.2-2mol/l,更优选0.2-1mol/l。当反应中单体的浓度超过1mol/l时,该反应会由于过高的支化程度而产生交联难以控制,所得聚合物难以溶解于有机溶剂中;而当反应浓度低于1mol/l时,则可以形成线型或支化结构的聚合物。
优选地,所述催化剂选自金属有机催化剂。
优选地,所述金属有机催化剂包括含钌、钼或钨的金属有机催化剂中的至少一种。所述用于本发明的催化剂具有优异的烯烃复分解催化能力。
优选地,所述环状烯烃单体与催化剂的浓度比为200-4000:1。当反应单体和催化剂之间比例达到1000:1或更高时,反应的速率较慢,支化程度较低,所得分子量较高,分布较窄;当反应物和催化剂之间比例达到500:1或更低时,反应的速率较快,支化程度较高,所得分子量较低,分布较宽。
优选地,所述聚合反应的温度0-50℃,反应时间为0.5-72h。随反应温度的升高,反应的支化程度增加,例如,当反应温度为50℃时,所得聚合物的支化程度可以达到30.7%,呈超支化的结构;而当反应温度为27℃时,所得聚合物的支化程度仅为5.3%,呈线型结构。
更优选地,所得聚合物为超支化结构时,聚合反应的温度控制在50℃,反应时间为72h;所得聚合物为线型结构时,聚合反应的温度控制在0℃,反应时间为3h。
优选地,所述聚合反应在有机溶剂存在下进行,所述有机溶剂包括四氢呋喃、丙酮、甲苯、氯仿、二氯甲烷中的至少一种。
本发明还提供了一种环状烯烃单体,所述环状烯烃单体如式i所示:
本发明还提供了一种环状烯烃单体在合成支化程度可控的聚烯烃材料中的应用。
本发明的方法得到的产物中相邻两个叔碳间碳原子的个数由单体之中参加复分解反应的两个不饱和碳官能团之间碳原子的个数决定;当反应温度升高时,产物的支化程度升高,反之亦然;当反应的浓度提高时,产物的分子量会升高,分子量分布变宽,反之亦然;当反应的单体浓度提高时,支化程度升高、分子量升高,反之亦然;当反应的催化剂含量提高时,产物的分子量升低、分布变宽、支化程度提高,反之亦然;当反应时间提升时,产物的分子量升高、分布变宽、支化程度提高。
与现有技术相比,本发明具有如下的有益效果:
本发明通过特定设计一系列具有环状烯烃单体,并利用这些单体进行聚合反应形成聚合物,通过改变反应单体和反应参数如反应温度、溶剂种类、催化剂浓度、单体浓度和反应时间等,可实现对聚烯烃支化程度、分子量及其分布的控制,由此可以制备支化结构精确可控的超支化聚烯烃。
根据本发明制备的聚烯烃材料支化程度可控、粘度低、流动性好,可广泛应用于涂料、润滑剂、聚合物加工流动改善剂等领域。
附图说明
通过阅读参照以下附图对非限制性实施例所作的详细描述,本发明的其它特征、目的和优点将会变得更明显:
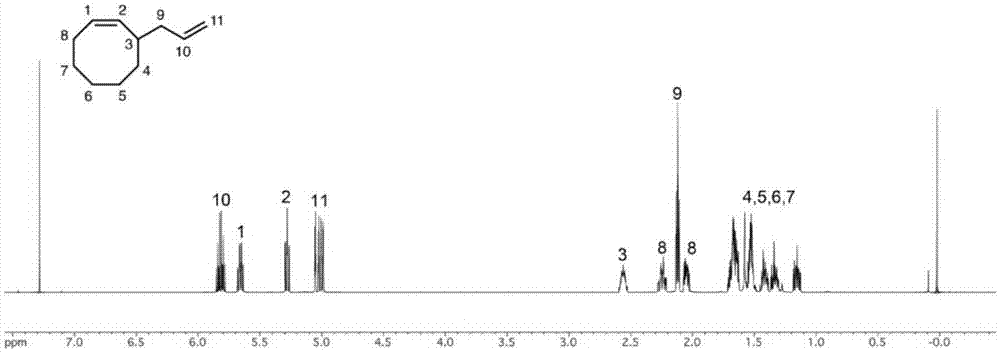
图1为实施例1中合成的3-乙烯基-1-环辛烯的结构及其核磁共振氢谱;
图2为实施例2至5中合成的3-烯丙基-1-环辛烯的结构及其核磁共振氢谱;
图3为实施例6中合成的侧基具有8个碳原子的环辛烯衍生物的结构及其核磁共振氢谱;
图4为实施例1中单体经聚合反应后所得产物的支化结构及其核磁共振氢谱,其各峰的积分值会因反应条件不同而发生变化;
图5为实施例2至5中单体经聚合反应后所得产物的支化结构及其核磁共振氢谱,其各峰的积分值会因反应条件(实施例)不同而发生变化;
图6为实施例6中合成的单体经聚合反应后所得产物的支化结构及其核磁共振氢谱,其各峰的积分值会因反应条件不同而发生变化;
图7为实施例1中合成的聚合物的支化结构。
具体实施方式
下面结合具体实施例对本发明进行详细说明。以下实施例将有助于本领域的技术人员进一步理解本发明,但不以任何形式限制本发明。应当指出的是,对本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变化和改进。这些都属于本发明的保护范围。
实施例1
单体合成:取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入浓度为1mol/l的乙烯基溴化镁四氢呋喃溶液,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水出水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ia所示,产率约为40%。核磁共振氢谱如图1所示。
聚合反应:取上述步骤所得纯净单体91mg,加入无水四氢呋喃定容在880μl(0.75mol/l),置于50℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(2.7mg催化剂溶于25ul四氢呋喃中,3.7mmol/l)加入到该反应中,保持反应密闭进行48h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。
如图4所示,其数均分子量为0.89kda,重均分子量为3.92kda,分子量分布为4.4,支化度为9.1%。
实施例2
单体合成:取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入浓度为1mol/l的烯丙基溴化镁四氢呋喃溶液,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水出水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ib所示,产率约为37%。核磁共振氢谱如图2所示。
聚合反应:取上述步骤所得纯净单体100mg,加入无水四氢呋喃定容在880μl(0.75mol/l),置于50℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(2.7mg催化剂溶于25ul四氢呋喃中,3.7mmol/l)加入到该反应中,保持反应密闭进行24h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。该聚合物的支化结构与式iia类似,其主链结构与末端单元如图5所示,其数均分子量为14.4kda,重均分子量为86.2kda,分子量分布为6.0,支化度为30.7%。该聚合物的支化结构如如7所示。
实施例3
单体合成:取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入浓度为1mol/l的乙烯基溴化镁四氢呋喃溶液,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水出水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ib所示,产率约为40%。
聚合反应:取上述步骤所得纯净单体100mg,加入无水四氢呋喃定容在330μl(1mol/l),置于27℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(2.7mg催化剂溶于25ul四氢呋喃中,3.7mmol/l)加入到该反应中,保持反应密闭进行24h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。该聚合物的支化结构与图7相类似,其主链结构与末端单元如图5所示,其数均分子量为38.1kda,重均分子量为131.5kda,分子量分布为3.4,支化度为15.4%。
实施例4
单体合成:取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入浓度为1mol/l的乙烯基溴化镁四氢呋喃溶液,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水出水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ib所示,产率约为40%。
聚合反应:取上述步骤所得纯净单体100mg,加入无水四氢呋喃定容在880μl,(0.75mol/l)置于50℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(0.135mg催化剂溶于25ul四氢呋喃中,0.18mmol/l)加入到该反应中,保持反应密闭进行24h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。该聚合物的支化结构与图7相类似,其主链结构与末端单元如图5所示,其数均分子量为2.6kda,重均分子量为4.8kda,分子量分布为1.8,支化度为15.4%。
实施例5
单体合成:取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入浓度为1mol/l的乙烯基溴化镁四氢呋喃溶液,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水出水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ib所示,产率约为40%。
聚合反应:取上述步骤所得纯净单体100mg,加入无水四氢呋喃定容在880μl(0.75mol/l),置于50℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(2.7mg催化剂溶于25ul四氢呋喃中,3.7mmol/l)加入到该反应中,保持反应密闭进行6h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。该聚合物的支化结构与图7相类似,其主链结构与末端单元如图5所示,其数均分子量为6.5kda,重均分子量为22.2kda,分子量分布为3.4,支化度为27.5%。
实施例6
单体合成:取镁粉3g,加入30ml无水四氢呋喃中,在氩气保护下加入8-溴-1-辛烯溶液(12g溶于30ml无水四氢呋喃中),待格式反应开始后,维持反应在室温下30min,而后加热到50℃下维持1小时,得到格式试剂。
取3-溴环辛烯10g,加入到30ml无水四氢呋喃中,在氩气保护下缓慢搅拌并加入碘化亚铜0.1g,随后在0℃下缓慢滴入本实施例中制备的格式试剂,反应3小时。向反应液中缓慢加入饱和氯化铵溶液以终止反应,将反应液用乙醚萃取三次,收集有机相并用饱和食盐水除水、加入无水硫酸镁干燥,而后用旋转蒸发浓缩反应液,得到粗产物并使用硅胶柱进行纯化(流动相为正己烷),得到纯净产物,结构如式ic所示,产率约为49%。核磁共振氢谱如图3所示。
聚合反应:取上述步骤所得纯净单体147mg,加入无水四氢呋喃定容在880μl(0.75mol/l),置于50℃下缓慢搅拌(搅拌速率为100rpm),而后在将grubbs2ndgeneration催化剂的溶液(2.7mg催化剂溶于25ul四氢呋喃中,3.7mmol/l)加入到该反应中,保持反应密闭进行24h,而后加入500ul的乙烯基乙醚终止反应,采用甲醇沉淀三次,得到纯净的聚合物。该聚合物的支化结构与式iia相类似,其主链结构与末端单元如图6所示,其数均分子量为102kda,重均分子量为534kda,分子量分布为5.23,支化度为15.6%。
需要说明的是,作为以上实施例的替换方案,当n为大于等于1的其他值时,同样可以制备得到支化程度可控的式iia结构。
以上对本发明的具体实施例进行了描述。需要理解的是,本发明并不局限于上述特定实施方式,本领域技术人员可以在权利要求的范围内做出各种变化或修改,这并不影响本发明的实质内容。在不冲突的情况下,本申请的实施例和实施例中的特征可以任意相互组合。
- 还没有人留言评论。精彩留言会获得点赞!