化合物、组合物及其在药物制备中的用途的制作方法
本发明涉及雄激素受体的新型拮抗剂化合物;涉及在治疗与雄激素受体相关的疾病,如前列腺癌,使用这类化合物的方法;和涉及包含这些化合物的药物组合药。
背景技术
雄激素受体(androgenreceptor,ar)是一个配体依赖性的转录调节蛋白,属于核受体超家族成员。雄激素受体和雄激素,比如睾硐或者更有效的双氢睾硐(dth)结合,导致构象变化,引起雄激素受体活化。活化的雄激素受体以二聚体形式与靶细胞核中特定的dna序列-雄激素反应元件(are)结合,调节靶基因的表达,对细胞的增殖、生存和分化产生相应的生物效应。雄性激素受体在许多男性荷尔蒙相关的疾病中扮演重要角色,例如前列腺癌等。前列腺癌对雄激素有较高的敏感性,前列腺癌细胞有雄激素受体高表达的特征。
前列腺癌是男性生殖系统最常见的恶性肿瘤,许多年纪大的男性最终均出现显微症状的前列腺癌。与其它恶性肿瘤相比较,前列腺癌的生长相当缓慢,90%的前列腺癌患者几十年保持潜伏状态,无明显临床症状。象许多其它癌症一样,初期如不进行治疗,不断发展,最后会经血液或淋巴液传输到其它组织。
目前,雄激素受体拮抗剂(arantagonist)是治疗前列腺癌的主要化学药物,已被批准临床使用的雄激素受体拮抗剂(arantagonist)(如比卡鲁胺、恩杂鲁胺、apalutamide)在临床治疗应用上可以减慢前列腺癌的发展,其作用为直接阻止睾硐或双氢睾酮(dht)与雄激素受体结合,从而阻断雄激素的生理作用。不过,上述这几个药物直接杀死前列腺癌细胞的能力不显著,细胞生物学实验中,其ic50在10um以上。
因此,本领域仍需要继续寻找出对高表达雄激素受体的前列腺癌细胞具有选择性杀死能力的化合物,来攻克激素难治的前列腺癌,避免或者减缓激素敏感的前列腺癌的发展。
技术实现要素:
根据本发明,提出一种调节核激素受体,尤其雄激素受体的功能的化合物。这些化合物可导致前列腺癌纽胞和肿瘤的消失。
本发明的化合物具有式i的结构:
其中,r1选自氢、氟、氯;r2和r3独立地选自氢、烷基、取代的烷基、链烯基或取代的链烯基、炔基或取代的炔基、芳基,r2和r3可连接以形成环,所述环可为环烷基、取代的环烷基、芳香杂环或非芳香杂环;r4选自氢、氰基、烷基、取代的烷基、链烯基或取代的链烯基、炔基或取代的炔基、芳基;r5选自氢、卤素、卤代烷基。
在一种具体实施方式中,r1为氢或氟。
在一种具体实施方式中,r2和r3独立地为c1-c3的烷基,或者合起来形成一个c3-c6环烷基。
在一种具体实施方式中,r4为氰基。
在一种具体实施方式中,r5为三卤甲烷基,更优选为三氟甲基。
在本发明的合成实施例中,合成了下述化合物:
4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺
4-(3-(6-氰基-5-(三氟甲基)吡啶-3-基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺
4-(3-(3-氯-4-氰基苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺
4-(7-(4-氰基-3-(三氟甲基)苯基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺
4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺
在实施方案中,药物组化合物包括治疗有效剂量的式i的化合物或其可药用盐,和可药用载体、稀释剂或助剂。
药物组合物可包括二甲亚砜、羧甲纤维素、聚山梨酯和水的溶液。药物组合物可包括二甲亚砜、羧甲纤维素、聚山梨酯和水。
方法的实施方案包括预防或治疗过度增生病症如激素敏感的前列腺癌或激素难治的前列腺癌的方法,可包括向有此需要的给药根据式i的化合物或其可药用盐,从而预防或治疗过度增生病症。化合物可以通过紧密注射给药、通过注射入组织给药、腹膜内给药、口服给药。
在实施方案中,式i化合物与雄激素受体(androgenreceptor,ar)有强的亲和力,可抑制ar通路下游蛋白的表达。式i化合物有明显组蛋白脱乙酰基酶抑制活性。对雄激素受体高表达的肿瘤细胞增殖有明显抑制作用。
附图说明
图1:药物与ar-lbd结合的ic50结果图示
图2:药物的荧光素酶报告基因实验结果的曲线图。
图3:药物在vcap细胞中对ar激活的下游蛋白的表达结果。
图4:药物对tubulin蛋白乙酰化的影响。
图5:药物对组蛋白h3乙酰化的影响。
图6:药物对vcap细胞的增殖抑制作用。
图7:处理4天后zeta55对三种细胞的增殖抑制的选择性。
具体实施方式
本发明的化合物具有式i的结构:
其中,r1选自氢、氟、氯;r2和r3独立地选自氢、烷基、取代的烷基、链烯基或取代的链烯基、炔基或取代的炔基、芳基,r2和r3可连接以形成环,所述环可为环烷基、取代的环烷基、芳香杂环或非芳香杂环;r4选自氢、氰基、烷基、取代的烷基、链烯基或取代的链烯基、炔基或取代的炔基、芳基;r5选自氢、卤素、卤代烷基。
本文所用的术语“烷基”表示支链或直链的烃链,优选具有约1至约5个碳,如甲基、乙基、正丙基、异丙基、正丁基、仲丁基、异丁基、叔丁基、2-甲基戊基、戊基等。“取代的烷基”是指前述烷基被一个或多个官能团如羟基、溴、氟、氯、碘、巯基或硫代、氰基、烷基硫代、杂环基、芳基、杂芳基、羧基、烷氧羰基(carbalkoyl)、烷基、链烯基、硝基、氨基、烷氧基、酰氨基等任选取代以形成如三氟甲基、3-羟基己基、2-羧基丙基、2-氟乙基、羧甲基、氰基丁基等的烷基。
除非特别说明,本文单独或作为另一基团的部分所使用的术语“环烷基”包括包含1至2个环的饱和或部分不饱和(包含1或多个双键)环状烃基团,优选包括3至10个碳,例如环丙基、环丁基、环戊基、环己基、环庚基、环辛基和环癸基。“取代的环烷基”包括环烷基,其被一个或多个取代基如卤素、烷基、烷氧基、羟基、芳基、芳氧基、芳基烷基、环烷基、烷基酰氨基、烷酰基氨基、氧代、酰基、芳基羰基氨基、氨基、硝基、氰基、硫醇和/或烷基硫代和/或包括在“取代的烷基”定义中的任何取代基任选取代。
除非特别说明,本文单独或作为另一基团的部分所使用的术语“链烯基”是指在主链(normalchain)中有2至10个碳、优选2至8个碳和更优选2至5个碳的在主链中包括一个或多个双键的直链或支链基团,如乙烯基、2-丙烯基、3-丁烯基、2-丁烯基、4-戊烯基、3-戊烯基、2-己烯基、3-己烯基、2-庚烯基、3-庚烯基、4-庚烯基、3-辛烯基、3-壬烯基、4-癸烯基。“取代的链烯基”包括链烯基,其用一个或多个取代基如以上在定义“取代的烷基”和“取代的环烷基”中所包括的取代基任选取代。
除非特别说明,本文单独或作为另一基团的部分所使用的术语“炔基”是指在主链中有2至8个碳且在主链中包括一个或多个三键的直链或支链基团,如2-丙炔基、3-丁炔基、2-丁炔基、4-戊炔基、3-戊炔基、2-己炔基、3-己炔基、2-庚炔基、3-庚炔基、4-庚炔基、3-辛炔基。“取代的炔基”包括炔基,其用一个或多个取代基如以上在定义“取代的烷基”和“取代的环烷基”中所包括的取代基任选取代。
除非特别说明,本文中单独或作为另一基团的部分使用的术语“芳基”或“ar”是指在环部分中包含6至10个碳的单环和多环芳香基团(如苯基或萘基,包括1-萘基和2-萘基)和可任选地包括稠合至碳环或杂环(如芳基、环烷基、杂芳基或环杂烷基环)上的一个至三个额外的环。
“取代的芳基”包括用一个或多个官能团任选取代的上述芳基,该官能团如卤代、烷基、卤代烷基、烷氧基、卤代烷氧基、链烯基、三氟甲基、三氟甲氧基、炔基、环烷基、环烷基烷基、羟基、硝基、氰基、氨基(其中氨基包括1或2个烷基取代的氨基)。
除非另有所指,本文所用的术语“杂环”或“杂环”表示未取代的或取代的稳定5-至10-元单环体系,其可为饱和或不饱和的,由碳原子和选自n、o或s的1至4个杂原子组成,且其中氮和硫杂原子可任选地被氧化,且氮杂原子可任选地被季铵化。这种杂环基团的例子包括,哌啶基、哌嗪基、氧哌嗪基、吡咯基、吡咯烷基、呋喃基、噻吩基、吡唑基、吡唑烷基、咪唑基。
式i的化合物可作为可药用盐存在,这也在本发明的范围内。如果式i化合物具有例如至少一个碱性中心,它们可形成酸加成盐。这些例如使用强无机酸、强有机羧酸或有机磺酸形成,该强无机酸如矿物酸例如硫酸、磷酸或氢卤酸,该强有机羧酸如未取代的或取代(例如被卤素取代)的1至4个碳原子的烷烃羧酸例如乙酸、如饱和或不饱和二羧酸例如草酸、丙二酸、琥珀酸、马来酸、富马酸、邻苯二甲酸或对苯二甲酸、如羟基羧酸例如抗坏血酸、乙醇酸、乳酸、苹果酸、酒石酸或柠檬酸、如氨基酸(例如天冬氨酸或谷氨酸或赖氨酸或精氨酸)、或苯甲酸,该有机磺酸如未取代的或取代(例如被卤素取代)的(c1-c4)烷基或芳基磺酸例如甲基或对甲苯-磺酸而形成。如果需要,也可额外衍生一个碱性中心以形成相应的酸加成盐。
本发明化合物可以药物组合物的形式使用,其中包含治疗有效量的本文所限定的本发明化合物和可药用载体或稀释剂。
可以调节根据本发明的药物组合物的形式,以适于用多种给药途径向需要治疗的患者例如哺乳动物如人患者给药,例如口服给药、鼻内给药、腹膜内给药、或非消化道给药、通过静脉内、肌内、局部或皮下路径给药、或通过注射入组织给药。这种组合物和制剂应该包含至少0.01%的一种或多种本发明化合物。组合物和制剂的百分数当然可以变化和可以例如是给定单元剂型的约0.05%至约2%重量。化合物在这种治疗有用的组合物中的量使得获得有效的剂量水平。
本发明化合物可全身给药,如口服、与可药用载体如惰性稀释剂或可同化的食用载体组合、或通过吸入或吹入。它们可被包封在硬或软壳胶囊中、可被压成片剂、或可与病人食用的食品直接混合。对于口服治疗给药,本发明化合物可与一种或多种赋形剂组合和以可摄取的片剂、口含片剂、锭剂、胶囊、酏剂、悬浮液、糖浆、干胶片(wafer))等的形式使用。该化合物可与细的惰性粉状载体组合和由患者吸入或吹入。这种组合物和制剂应该包含至少0.1%的一种或多种本发明化合物。
片剂、锭剂、丸剂、胶囊等也可包含:粘合剂如西黄蓍胶、阿拉伯胶、玉米淀粉或明胶;赋形剂如磷酸二钙;崩解剂如玉米淀粉、马铃薯淀粉、藻酸等;润滑剂如硬脂酸镁;和甜味剂如蔗糖、果糖、乳糖或阿司帕坦,或可加入芳香剂如薄荷、冬青油或樱桃调味剂。当单元剂型是胶囊时,除了以上类型的材料,它还可包含液体载体如植物油或聚乙二醇。各种其它材料可存在作为涂层或以其它方式改变固体单元剂型的外形(physicalform)。例如,片剂、丸剂或胶囊可涂有明胶、蜡、虫胶、糖等。糖浆或酏剂可包含活性化合物、作为甜味剂的蔗糖或果糖、作为防腐剂的对羟基苯甲酸甲酯和对羟基苯甲酸丙酯、染料、和调味剂如樱桃或橙子调味剂。当然,用于制备任何单元剂型的任何材料在用量上应该是可药用的和实质上无毒的。另外,本发明化合物可引入持续释放制剂和设备。例如,化合物可引入延时释放(timerelease)胶囊、延时释放片剂和延时释放丸剂。
本发明化合物也可通过输注或注射而静脉内或腹膜内给药。化合物的溶液可在水中制备,任选地与非毒性表面活性剂混合。适用于注射或输注的药物剂型可包括无菌水溶液或分散体或无菌粉末。液体载体可以是溶剂或液体介质,包括例如水、乙醇、多元醇(例如甘油、丙二醇、液体聚乙二醇等)、植物油、非毒性甘油酯、和其合适的混合物。
对于局部给药,本发明化合物可以纯的形式使用。但是,通常期望将它们作为组合物或制剂与可以是固体或液体的皮肤学上可接受载体一起向皮肤给药。
有用的固体载体包括细分散的固体如滑石、粘土、微晶纤维素、硅石、矾土等。其它固体载体包括非毒性聚合物纳米颗粒或微颗粒。有用的液体载体包括水、醇或二醇或水/醇/二醇共混物,其中本发明化合物可在有效的水平下任选地借助于非毒性表面活性剂而溶解或分散。可加入助剂如香料和另外的抗微生物剂以针对给定用途而优化性能。所得液体组合物可由吸收剂垫施用、用于浸渍绷带和其它敷料、或使用泵-型或气溶胶喷雾器喷雾到受影响的区域上。
增稠剂如合成聚合物、脂肪酸、脂肪酸盐和酯、脂肪醇、改性的纤维素或改性的矿物材料也可与液体载体一起使用以形成可铺展的糊、凝胶、软膏、皂等,用于直接施用到使用者的皮肤上。
化合物在液体组合物如洗剂中的浓度可以是约0.1至约25%重量,或约0.5至约10%重量。在半固体或固体组合物如凝胶或粉末中的浓度可以是约0.1至约5%重量,或约0.5至约2.5%重量。
本发明化合物用于治疗所需的量不仅随着所选的特定盐变化,而且随着给药路径、正在治疗的病况的性质以及病人的年龄和病况而变化,且最终由主治医师或临床医师决定。
本发明试剂给药的有效剂量和路径是常规的。试剂的精确量(有效剂量)因患者不同而变化,取决于例如患者的种类、年龄、重量和一般或临床状态、正在治疗的任何病症的严重性或机理、所用的特定试剂或载体、给药的方法和进度等。治疗有效剂量可通过本领域技术人员已知的常规程序经验地确定。
一般来说,合适的剂量可以是约0.01至约500mg/kg/天,如约0.1至约500mg/kg体重/天,如约0.1至约100mg/千克受者体重/天。例如,合适的剂量可以是约1mg/kg、10mg/kg或50mg/kg体重/天。
本发明化合物便利地以单元剂型给药;例如,每单元剂型包含约0.0005至约500mg、约0.01至约50mg、约0.05至约10mg、或约5mg活性成分。
可给药本发明化合物以实现例如约0.5至约75μm、约1至50μm、约2至约30μm、或约5至约25μm的血浆浓度峰值。示例性的期望血浆浓度包括至少或不超过0.25、0.5、1、5、10、25、50、75、100或200μm。这可例如通过以下实现:静脉注射本发明化合物的0.05-5%溶液(任选地在盐水中),或以包含约1-1000mg该化合物的大丸剂口服给药。期望的血液水平可通过连续输注以提供约0.0005至约25mg/kg体重/小时,例如至少或不超过0.0005、0.005、0.05、0.5、5或25mg/kg/小时而保持。或者,这种水平可通过包含约0.002至约100mg/kg体重,例如至少或不超过0.002、0.02、0.2、2、20、50或100mg化合物/kg体重的间歇输注而得到。
本发明化合物可便利地以单剂量供给或作为在合适的间隔给药的分剂量例如作为2、3、4或更多子剂量(sub-dose)/天供给。子剂量自身可进一步例如分成许多离散的松散分开的给药;如从吹入器多次吸入。
合成实施例1
4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺(化合物zeta32)的合成
(1)4-(2-羧基丙-2-基)氨基)-2-氟苯甲酸15的合成
在150ml双口烧瓶中依次加入化合物2-氟-4-溴苯甲酸(4.00g,18mmol)、2-甲基-2-氨基丙酸(2.78g,27mmol)、碳酸钾(6.67g,48mmol)、cui(0.45g,2.4mmol)、乙酰丙酮(1.44g,14.4mmol)、30mldmf、2ml水和3滴三乙胺,在氮气保护装置下,140℃反应24h,tlc监控反应(乙酸乙酯:石油醚=1:1),反应完毕后加入20ml水,加浓盐酸调节ph至4,用乙酸乙酯(30×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物加入20ml异丙醇加热溶解,冷却结晶,过滤得到化合物15,3.47g,棕色固体,收率80.1%。.
(2)2-氟-4-((1-甲氧基-2-甲基-1-氧代丙-2-基)氨基)苯甲酸甲酯16的合成
在150ml双口烧瓶中加入化合物15(5.00g,20.7mmol)和40ml甲醇,冰浴下滴入4.2ml(58mmol)氯化亚砜,随后升温至80℃回流搅拌,反应2小时,反应液冷却至室温,加入20%碳酸钠溶液调节溶液ph至8,乙酸乙酯(30×3ml)萃取,饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压抽除溶剂得化合物16,4.97g,棕色固体,收率95.6%。
(3)4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸甲酯17的合成
在50ml双口烧瓶中依次加入化合物16(1.00g,4.0mmol)、4-异硫氰基-2-三氟甲基苯甲腈(1g,57.6mmol)、1mldmso和10ml乙酸乙脂,85℃反应18h,tlc监控反应(石油醚:乙酸乙酯=3:1),反应完毕后加入10ml水,乙酸乙酯(20×3ml)萃取,饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压抽除溶剂,残余物加入10ml异丙醇加热溶解,冷却结晶,过滤得到化合物17,1.1g,淡黄色固体,收率60.1%。
(4)4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸19的合成
在150ml双口烧瓶中加入化合物17(3g,6.45mmol)、25ml甲醇和25ml1mol/l氢氧化钠溶液,20℃搅拌4h,反应完毕加入2mol/l稀盐酸调节至ph为4,用乙酸乙酯(50×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,得到化合物11,淡黄色固体,2.86g,收率98.2%。
(5)4-(3-(4-氰基-3-(三氟甲基)苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺20的合成
在25ml双口烧瓶中加入化合物19(0.5g,1.1mmol)、8mlthf和cdi(0.357g,2.2mmol),30℃搅拌10h后加入盐酸羟胺(0.230g,3.3mmol)在室温下反应10h,tlc监控反应(乙酸乙酯:石油醚:甲醇=20:5:2),反应完毕加8ml水,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经硅胶柱层析色谱纯化,得到化合物zeta32,0.144g,淡黄色固体,收率28.1%。
m.w.:466.41;1hnmr(500mhz,dmso-d6):δ11.16(s,1h),9.34(s,1h),8.41(d,j=10.0hz,1h),8.30(s,1h),8.09(d,j=5.0hz,1h),7.75(t,j=5.0hz,1h),7.43(d,j=5.0hz,1h),7.35(s,1h),2.51(s,6h);13cnmr(125mhz,dmso-d6):δ180.55,175.18,160.98,160.40,158.41,138.94,138.40,136.74,134.42,131.31,128.48,126.67,123.96,118.61,118.42,115.47,109.21,67.08,23.42.
合成实施例2
4-(3-(6-氰基-5-(三氟甲基)吡啶-3-基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺(化合物zeta33)的合成
(1)4-(3-(6-氰基-5-(三氟甲基)吡啶-3-基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸甲酯25的合成
在25ml双口烧瓶中加入化合物21(0.60g,2.5mmol)、3-氯-4-氰基苯胺(0.561g,3mmol)、8mln,n-二甲基乙酰胺和硫光气(0.35ml,3.8mmol),升温至70℃反应24h,冷却至室温后加入4ml甲醇、1ml浓盐酸和2ml水,回流搅拌2h,冷却至室温后,乙酸乙酯(20×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经柱层析纯化得到化合物25,0.355g,黄色固体,收率30.5%。
(2)4-(3-(6-氰基-5-(三氟甲基)吡啶-3-基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸26的合成
在25ml双口烧瓶中将化合物25(0.230g,0.5mmol)溶于2ml甲醇,往此混合物中加入2ml1mol/l氢氧化钠,20℃搅拌4h,反应完毕后加入2mol/l稀盐酸调节至ph为4),用乙酸乙酯(20ml×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,得到化合物26,0.220g,淡黄色固体,收率98.1%。
(3)4-(3-(6-氰基-5-(三氟甲基)吡啶-3-基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺27的合成
往25ml双口烧瓶中依次加入化合物26(0.200g,0.44mmol)、4mlthf和cdi(0.143g,0.88mmol)于30℃搅拌10h随后加入盐酸羟胺(0.092g,1.32mmol),在室温下反应10h,tlc监控反应(乙酸乙酯:石油醚:甲醇=20:5:2)反应完毕加4ml水,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经硅胶柱层析色谱纯化,得化合物a8,0.064g,淡黄色固体,收率31.6%。
m.w.:467.40;1hnmr(500mhz,cdcl3):δ8.95(d,j=1.0hz,1h),8.46(d,j=2.0hz,1h),7.85(t,j=9.0hz,1h),7.38(t,j=9.5hz2h),1.61(s,1h);13cnmr(125mhz,cd3od):δ180.63,175.26,162.16,158.77,151.31,139.59,135.87,130.85,126.25,123.76,112.21,122.09,118.34,118.15,66.78,22.34.
合成实施例3
4-(3-(3-氯-4-氰基苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺(zeta34)的合成
(1)4-((2-氰基丙-2-基)氨基)-2-氟苯甲酸甲酯21的合成
在150ml双口烧瓶中于室温下依次加入2-氟-4-氨基苯甲酸脂2(4.5g,26.6mmol)、丙酮(2.99g,42.7mmol)、氰化钾(2.6g,40mmol)和46ml醋酸,将此混合液升温至75℃反应24h,tlc监控反应(石油醚:乙酸乙酯=20:3),反应完毕后往反应液中加入20ml水,乙酸乙酯萃取(40×3ml),减压抽除溶剂,残余物加入6ml乙醇和5ml水的混合溶剂加热溶解,冷却结晶,过滤得化合物21,3.13g,黄色固体,收率50.2%。
(2)将4-(3-(3-氯-4-氰基苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸甲酯22的合成
在25ml双口烧瓶中加入化合物21(0.500g,2.1mmol)、3-氯-4-氰基苯胺(0.38g,2.5mmol)和8mln,n-二甲基乙酰胺,往此混合液中滴入硫光气(0.25ml,3.89mmol),升温至95℃反应24h,冷却至室温后加入2ml甲醇、0.5ml浓盐酸和2ml水,回流搅拌2h,冷却至室温后,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经柱层析纯化得到化合物22,0.257g黄色固体,收率28.8%。
(3)将4-(3-(3-氯-4-氰基苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟苯甲酸23的合成
在25ml双口烧瓶中加入化合物22(0.120g,0.28mmol)、1ml甲醇和1ml1mol/l氢氧化钠溶液,20℃搅拌4h,反应完毕加入2mol/l稀盐酸调节至ph至4,用乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,得到化合物23,0.140g,淡黄色固体,收率98.2%。
(4)4-(3-(3-氯-4-氰基苯基)-5,5-二甲基-4-氧代-2-硫代咪唑烷-1-基)-2-氟-n-羟基苯甲酰胺24的合成
在25ml双口烧瓶中加入化合物23(0.120g,0.29mmol)、4mlthf和cdi(0.093g,0.58mmol),于30℃下搅拌10h后加入盐酸羟胺(0.060g,0.87mmol),在室温下反应10h,tlc监控反应(乙酸乙酯:石油醚:甲醇=20:5:2),反应完毕加4ml水,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经硅胶柱层析色谱纯化得化合物a7,0.038g,淡黄色固体,收率30.5%。m.w.,432.85;1hnmr(500mhz,cd3od):δ7.97(t,j=7.5hz,1h),7.82(d,j=9.0hz1h),7.67(s,1h),7.65(dd,j=1.5hz,1h),7.37(t,j=10.0hz,2h),1.60(s,6h);13cnmr(125mhz,cd3od):δ180.46,175.10,162.24,160.76,138.53,136.14,134.12,130.77,130.39,128.13,126.16,122.47,119.94,118.30,114.87,113.02,66.57,22.32.
合成实施例4
4-(7-(4-氰基-3-(三氟甲基)苯基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺(化合物zeta55)的合成
(1)2-氟-4-氨基苯甲酸甲酯2的合成
在100ml双口烧瓶中加入2-氟-4-氨基苯甲酸1(10.00g,64.5mmol)和30ml甲醇,冰浴下往此溶液中滴入6.7ml(92.0mmol)氯化亚砜,随后升温至回流,反应3小时,反应液冷却至室温后加入20%碳酸钠溶液调节溶液ph至8,乙酸乙酯(30×3ml)萃取,饱和氯化钠溶液洗涤,无水硫酸钠干燥,减压抽除溶剂得化合物2,10.36g,棕色固体,收率95.5%。
(2)4-((1-氰基环丁基)氨基)-2-氟苯甲酸甲酯3的合成
在250ml双口烧瓶中加入2-氟-4-氨基苯甲酸甲酯2(3.70g,21.8mmol)、环丁酮(2.44g,34.8mmol)、氰化钾(2.12g,32.6mmol)和37ml醋酸,升温至80℃反应12h,tlc监控反应,(石油醚:乙酸乙酯=20:3),反应完毕后往反应液中加入20ml水,乙酸乙酯萃取(30×3ml),减压抽除溶剂得化合物3,5.01g,棕红色固体,收率92.5%。
(3)(7-(4-氰基-3-(三氟甲基)苯基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟苯甲酸甲酯4的合成
在25ml双口烧瓶中加入化合物3(0.485g,1.95mmol)、3-三氟甲基-4氰基苯胺(0.435g,2.34mmol)和6mln,n-二甲基乙酰胺,待搅拌溶解后滴入硫光气(0.25ml,2.96mmol),升温至70℃反应24h,冷却至室温后加入2ml甲醇、0.5ml浓盐酸和2ml水,回流搅拌2h,冷却至室温后,用乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经柱层析纯化得到化合物5,0.281g,黄色固体,收率30.2%。
(4)(7-(4-氰基-3-(三氟甲基)苯基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟苯甲酸5的合成
在25ml双口烧瓶中加入化合物4(0.30g,0.62mmol)和2.5ml甲醇,往此混合物中加入2.5ml1mol/l氢氧化钠溶液,20℃下搅拌4h,反应完毕后加入2mol/l稀盐酸调节至ph至4,用乙酸乙酯(20×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,得到化合物5,0.281g,淡黄色固体,收率98.2%。
(5)4-(7-(4-氰基-3-(三氟甲基)苯基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺6的合成
在25ml双口烧瓶中依次加入化合物5(0.181g,0.4mmol)、cdi(0.130g,0.8mmol)和10mlthf,在30℃下搅拌10h后,加入盐酸羟胺(0.083g,1.2mmol)在25℃下反应10h,tlc监控反应,(乙酸乙酯:石油醚:甲醇=20:5:2)反应完毕后加5ml水,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经硅胶柱层析色谱纯化,得化合物a5,0.579g,淡黄色固体,收率30.3%。m.w.,478.42;1hnmr(500mhz,cd3od):δ8.15(d,j=5.0hz,2h),7.98–7.96(m,1h),7.88(t,j=5.0hz,1h),7.39(t,j=5.0hz,2h),2.71–2.66(m,2h),2.59–2.53(m,2h),2.15–2.09(m,1h),1.66–1.60(m,1h)13cnmr(125mhz,cd3od):δ180.48,175.00,162.17,158.90,139.70,137.98,135.39,133.42,132.98,131.01,127.34,126.70,118.74,118.54,114.59,109.28,108.91,67.74,31.19.
合成实施例5
4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺(zeta57)。
(1)4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟苯甲酸甲酯10的合成
在25ml双口烧瓶中加入化合物3(0.50g,2.0mmol)、3-三氟甲基-4氰基苯胺(0.448g,2.40mmol)和6.5mln,n-二甲基乙酰胺,往此溶液中滴入硫光气(0.28ml,3.20mmol),升温至70℃反应24h,冷却至室温后加入2ml甲醇、0.5ml浓盐酸和2ml水,回流搅拌2h,冷却至室温后,用乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经柱层析纯化得到化合物5,0.286g,淡黄色固体,收率29.9%。.
(2)4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟苯甲酸11的合成
在25ml双口烧瓶中加入化合物10(0.12g,0.25mmol)和1ml甲醇,往此混合物中加入1ml1mol/l氢氧化钠,20℃搅拌过夜,加入2mol/l稀盐酸调节至ph为4,乙酸乙酯(10×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,得到化合物11,0.113g,淡黄色固体,收率97.1%。
(3)4-(7-(6-氰基-5-(三氟甲基)吡啶-3-基)-8-氧代-6-硫代-5,7-二氮杂螺[3.4]辛烷-5-基)-2-氟-n-羟基苯甲酰胺12的合成
在25ml双口烧瓶中加入化合物12(0.097g,0.21mmol)、2mlthf和cdi(0.068g,0.42mmol)于30℃搅拌10h后,加入盐酸羟胺(0.044g,0.62mmol)在25℃下反应10h,tlc监控反应(乙酸乙酯:石油醚:甲醇=20:5:2),反应完毕加2ml水,乙酸乙酯(5×3ml)萃取,无水硫酸钠干燥,减压抽除溶剂,残余物经硅胶柱层析色谱纯化,得到化合物a6,0.031g,淡黄色固体,收率31.2%。m.w.,479,41;1hnmr(500mhz,cd3od):δ8.93(s,1h),8.42(s,1h),7.88(t,j=7.0hz,1h),7.40(t,j=10.5hz,2h),2.69(s,2h),2.56(d,j=10.0hz,2h),2.15–2.10(d,1h),1.63(d,j=9.5hz1h);13cnmr(125mhz,cd3od):δ180.74,175.18,162.16,158.91,151.26,150.86,149.36,139.70,135.74,131.03,126.67,125.74,122.37,122.24,118.70,118.51,67.86,31.19,13.14。
下面描述根据本发明的部分化合物的生物活性测试结果:
所用试剂:
dht(二氢睾酮),美国sigma公司提供
arn509,发明人合成
mdv3100,发明人合成。
1、ar拮抗活性检测
1.1拮抗剂与ar的结合实验
采用polarscreentmandrogenreceptor(ar)competitorassay,green试剂盒(美国lifetechnologies公司)检测拮抗剂与ar-lbd(gst)的亲和力。实验原理是ar-lbd与小分子荧光酮示踪剂(fluormonetracer)相结合,加入拮抗剂会竞争性地结合ar-lbd从而替换出荧光酮示踪剂,替换出的荧光酮示踪剂会发生荧光偏振,荧光偏振值与替换出示踪剂数量成反比。
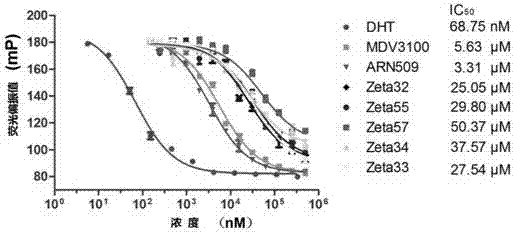
具体过程如下:将药物用dmso溶解成500um,然后在96孔板中两倍稀释浓度梯度至244nm,在384孔板中配置反应体系ar-lbd+tracer10ul加入10ul药物,阳性对照为ar-lbd+tracer10ul加入10uldht,锡箔纸包裹384孔板避光孵育6小时后在多功能酶标仪上读取荧光偏振值,用graphpadprism软件以药物浓度为横坐标,荧光偏振值为纵坐标绘制曲线。图1给出药物与ar-lbd结合的ic50结果图示。其中dht、mdv3100、arn509为阳性对照。
从图1中可以看出,合成的药物均可不同程度地与ar结合,其中zeta32、zeta33、zeta55的结合力最强,其ic50分别为25.05、27.54、29.80um。
1.2荧光素酶报告基因实验
用96孔板培养293t细胞,在293t细胞中共转染ar质粒和带有ar靶启动子的荧光素酶基因,ar激活靶启动子从而表达荧光素酶,荧光素酶的表达量与ar激活成正比。向细胞中加入dht可以激活ar,荧光素酶表达,检测的荧光信号增强。加入药物可通过检测荧光信号来测试其是否具有ar拮抗剂活性。
构建含有雄激素受体(ar)靶启动子的荧光素酶报告基因质粒mmtv-luc和表达雄激素受体(ar)的质粒psv-ar。将293t细胞接种到96孔板中,每孔1×104个细胞,孵育过夜使细胞贴壁,第二天采用
从图2可见,合成的药物对ar均有拮抗剂的作用,其拮抗能力从高到低依次为zeta32>zeta55>zeta34>zeta57>zeta33。。
1.3westernblot检测ar及psa表达
vcap细胞铺板于6孔板中,每孔接种细胞5×105个,孵育过夜使细胞贴壁,第二天每孔加入10um的药物,孵育24h后收集细胞,裂解细胞提取总蛋白,采用westernblot方法检测加入不同药物后细胞中psa的表达量,结果见图3。
从图3可以看出,合成的药物都能明显抑制ar通路下游蛋白(psa)的表达,提示合成的药物可以抑制ar的功能。
2、组蛋白脱乙酰基酶(hdac)抑制活性
用6孔板培养vcap细胞,vcap细胞加药处理24h后,用westernblot检测细胞质中tubulin和细胞核中h3蛋白质的乙酰化水平。实验过程如下:
vcap细胞铺板于6孔板中,每孔接种细胞5×105个,孵育过夜使细胞贴壁,第二天每孔加入10um的药物,孵育24h后收集细胞,裂解细胞提取细胞总蛋白,采用westernblot方法检测加入不同药物后细胞中kac-tubilin(定位细胞质)的表达量。图4给出实验结果,图中kac表示赖氨酸乙酰化。
vcap细胞铺板于6孔板中,每孔接种细胞5×105个,孵育过夜使细胞贴壁,第二天每孔加入10um的药物,孵育24h后收集细胞,采用epiquiktotalhistoneextractinkit(美国epigentek)提取组蛋白,采用westernblot方法检测加入不同药物后细胞中kac-h3(定位细胞核)的表达量。图5给出实验结果。
从图4、图5可以看出合成的药物都有一定的组蛋白脱乙酰基酶(hdac)抑制活性,可增强tubulin蛋白和组蛋白的乙酰化。对于细胞质中的tubulin乙酰化,zeta32、zeta55活性比较强(图4);而对于细胞核中的h3乙酰化,zeta32、zeta55、zeta57和zeta34活性比较强(图5)。
3、细胞增殖抑制活性检测
3.1药物对vcap细胞增殖的抑制作用
用96孔板培养雄激素敏感型前列腺癌细胞vcap细胞,vcap细胞在加有10nmdht的培养基中会增殖,我们通过加入药物处理4天以检测药物对前列腺癌细胞增殖的作用。
vcap细胞采用5%css和无酚红的1640培养基培养,接种于96孔板中,每孔加入细胞1×104个,孵育过夜使细胞贴壁,第二天加入不同浓度的药物,药物处理4天后采用mts试剂盒(美国promega)检测细胞活力,每孔加入10%的mts试剂,孵育2小时,然后在多功能酶标仪上检测490nm的吸光度值。用graphpadprism软件以药物浓度为横坐标,药物处理吸光度值/空白处理细胞吸光度值为纵坐标绘制曲线。
从图6可见,合成的药物对vcap细胞均有增殖抑制作用,抑制作用从高到低依次为zeta55>zeta32>zeta33>zeta34>zeta57。其中药物zeta55、zeta32和zeta33的细胞增殖抑制作用要高于阳性对照药物mdv3100。
3.2药物对细胞的选择性
293t细胞是ar阴性的人胚肾细胞,du145是ar阴性的前列腺癌细胞,vcap是ar阳性的前列腺癌细胞。用96孔板培养这三种细胞,分别加药(设浓度梯度)处理4天后mts检测药物对细胞增殖的抑制。具体过程如下:
vcap细胞采用5%css和无酚红的1640培养基培养,接种于96孔板中,每孔加入细胞1×104个。293t细胞和du145细胞采用10%fbs和1640培养基培养,接种于96孔板中,每孔加入细胞2×103个。孵育过夜使细胞贴壁,第二天加入不同浓度的药物,药物处理4天后采用mts试剂盒(美国promega)检测细胞活力,每孔加入10%体积的mts试剂,孵育2小时,然后在多功能酶标仪上检测490nm的吸光度值。用graphpadprism软件以药物浓度为横坐标,药物处理吸光度值/空白处理细胞吸光度值为纵坐标绘制曲线。
从图7可见,zeta55对ar阳性细胞vcap细胞有选择性的增殖抑制,zeta55对ar阳性细胞vcap的增殖抑制作用是ar阴性细胞(293t和du145)的20倍以上,提示zeta55可能具有ar阴性的细胞毒性较低。
综上所述,实验效果最好的药物是zeta55,其次是zeta32。zeta55的细胞增殖抑制作用好于已经上市的mdv3100和saha,而且对ar阳性细胞的作用远高于ar阴性细胞,提示该药物可能具有更好的疗效和更低的副作用。
- 还没有人留言评论。精彩留言会获得点赞!