一种配合物、其制备方法及应用了该配合物的催化剂与流程
本申请涉及一种二氧化碳还原阴极催化剂及其制备方法和应用,属于电催化技术领域。
背景技术
二氧化碳作为温室效应气体中重要的一种,通常产生于自然状况下的呼吸和人类的生产活动中。同时,对于许多的工业生产和地球上的植物来说,二氧化碳是一种非常重要的原料。在理想的情况下,地球上产生的二氧化碳和消耗应该达到一个平衡。然而,随着化石燃料所产生的二氧化碳不断增加。二氧化碳的平衡正在逐步被打破。因此,我们就要从源头上减少二氧化碳的排放以及将二氧化碳转化为有用的物质。
电催化就是这样一种在温和的反应条件下,将二氧化碳一步直接转化为一氧化碳、碳氢化合物和甲醇等高值化学品和液体燃料的方法。但同时二氧化碳电催化还原也存在着反应过电位较高以及反应过程中氢气的竞争反应导致产物的选择性低等挑战。因此,寻找一种高效的催化剂实现小的过电位以及较高的选择性和稳定性是很重要的。
技术实现要素:
根据本申请的一个方面,提供了一种配合物、其制备方法、应用了该配合物的二氧化碳还原阴极催化剂及其制备方法,该催化剂在有效的降低了二氧化碳还原反应过电位的同时,提高了催化剂选择性和稳定性。催化剂制备方法简单,选择性和稳定性高。
所述配合物具有式i所示的化学式:
l·2(h3o)·[pdcl4]·xh2o式i
其中,l代表瓜环类化合物分子;
x代表每摩尔配合物中游离水的摩尔数,6≤x≤8;
所述配合物属于单斜晶系,i2/c空间群,晶胞参数为
可选地,所述配合物晶体中,每摩尔配合物中包含瓜环空腔中一摩尔的游离水和瓜环外部六摩尔的游离水,每摩尔配合物含有一摩尔瓜环。
本申请的配合物晶体中,瓜环的空腔中心含有水分子,是在形成晶体的过程中生成的。原始加入的瓜环空腔并不含有水分子。
可选地,所述瓜环类化合物分子选自五元瓜环类化合物分子、六元瓜环类化合物分子中的至少一种。
可选地,所述瓜环类化合物分子为十甲基五元瓜环。
可选地,所述配合物的化学式为[me10cb[5]@h2o]·2h3o·[pdcl4]·6h2o,单斜晶系,i2/c空间群,
可选地,所述配合物的化学式为[me10cb[5]@h2o]·2h3o·[pdcl4]·6h2o,单斜晶系,i2/c空间群,
所述配合物为棕色晶体。
根据本申请的另一方面,提供一种配合物的制备方法,该方法操作简单,产率高。
所述的配合物的制备方法,其特征在于,以含有氯化钯的溶液i和含有瓜环类化合物的溶液ii为原料,采用溶液扩散法制备得所述配合物。
可选地,所述含有氯化钯的溶液i中还包括盐酸水溶液。
可选地,所述溶液扩散法采用h型扩散管,扩散时间不少于3小时;
溶液i中氯化钯的浓度为0.001mol/l~0.017mol/l;
溶液i中含有氢离子,氢离子的浓度为0.025mol/l~0.035mol/l;
溶液ii中瓜环类化合物的浓度为0.001mol/l~0.010mol/l。
可选地,溶液i中氯化钯的浓度的上限选自0.002mol/l、0.003mol/l、0.004mol/l、0.005mol/l、0.006mol/l、0.007mol/l、0.008mol/l、0.009mol/l、0.010mol/l、0.011mol/l、0.012mol/l、0.013mol/l、0.014mol/l、0.015mol/l、0.016mol/l或0.017mol/l;溶液i中氯化钯的浓度的下限选自0.001mol/l、0.002mol/l、0.003mol/l、0.004mol/l、0.005mol/l、0.006mol/l、0.007mol/l、0.008mol/l、0.009mol/l、0.010mol/l、0.011mol/l、0.012mol/l、0.013mol/l、0.014mol/l、0.015mol/l或0.016mol/l。
可选地,溶液i中氢离子的浓度的上限选自0.026mol/l、0.027mol/l、0.028mol/l、0.029mol/l、0.030mol/l、0.031mol/l、0.032mol/l、0.033mol/l、0.034mol/l或0.035mol/l;溶液i中氢离子的浓度的下限选自0.025mol/l、0.026mol/l、0.027mol/l、0.028mol/l、0.029mol/l、0.030mol/l、0.031mol/l、0.032mol/l、0.033mol/l或0.034mol/l。
可选地,溶液ii中瓜环类化合物的浓度的上限选自0.002mol/l、0.003mol/l、0.004mol/l、0.005mol/l、0.006mol/l、0.007mol/l、0.008mol/l、0.009mol/l或0.010mol/l;溶液ii中瓜环类化合物的浓度的下限选自0.001mol/l、0.002mol/l、0.003mol/l、0.004mol/l、0.005mol/l、0.006mol/l、0.007mol/l、0.008mol/l或0.009mol/l。
本领域技术人员可根据具体需要,选择合适的晶化时间。优选地,所述溶液扩散法采用h型扩散管,扩散时间不少于3小时。可选择地,扩散时间的范围下限选自3小时、5小时、10小时、15小时、20小时、2天、3天。进一步优选地,所述扩散时间为3小时~20天。更进一步优选地,所述扩散时间为3小时~14天。当扩散时间超过14天后,有杂晶生成。
可选地,所述方法包括:
(1)向氯化钯中加入去离子水和盐酸形成氯化钯的透明水溶液,即溶液i;
(2)在搅拌条件下,将me10cb[5]溶解在去离子水中,得到溶液ii;
(3)将溶液i和溶液ii分别转移至反应装置的一侧,扩散,得到所述配合物。
可选地,溶液i中氯化钯的用量为0.17mmol,6m盐酸溶液的用量为1ml,去离子水的用量为20ml。
可选地,溶液ii中me10cb[5]的用量为0.1mmol,去离子水的用量为20ml。
作为一种具体的实施方式,所述配合物的制备方法包括:
(1)向氯化钯中加入去离子水和盐酸形成氯化钯的透明黄色水溶液(溶液ⅰ)。
(2)在磁力搅拌下将me10cb[5]溶解在去离子水中(溶液ⅱ)。
(3)将溶液ⅰ与ⅱ分别小心地转移到h管的每一侧。在h管中,通过缓慢扩散三天后获得棕色晶体。
根据本申请的再一方面,提供一种纳米材料,其特征在于,所述纳米材料为配合物还原后得到;所述配合物选自所述的配合物、根据所述方法制备得到的配合物中的至少一种。
所述纳米材料为瓜环包裹的纳米钯。
可选地,所述纳米材料应用于电催化二氧化碳还原反应。
可选地,所述配合物的还原方法包括:250~350℃氢化1~10小时。
可选地,所述配合物为通过扩散法长成的晶体;所述配合物的还原为氢气将二价pd缓慢的还原,在还原的过程中,利用超分子组装中的氢键以及瓜环的限域来减弱纳米pd的团聚,更充分的发挥其对co2rr的催化性能。
可选地,所述氢化的温度的上限选自260℃、270℃、280℃、290℃、300℃、310℃、320℃、330℃、340℃或350℃;所述氢化的温度的下限选自250℃、260℃、270℃、280℃、290℃、300℃、310℃、320℃、330℃或340℃。
可选地,所述氢化的时间的上限选自2小时、3小时、4小时、5小时、6小时、7小时、8小时、9小时或10小时;所述氢化的时间的下限选自1小时、2小时、3小时、4小时、5小时、6小时、7小时、8小时或9小时。
可选地,所述配合物的还原方法包括:将配合物在氢气气氛下,250~350℃加热1~10小时,得到所述纳米材料;
其中,所述氢气气氛的组成为:体积分数为5%的氢气和体积分数为95%的氮气。
可选地,所述纳米材料的制备方法包括,将配合物研磨后,通入(5%氢气+95%氮气),250~350℃加热1~10小时,还原得到所述纳米材料;
所述配合物选自所述的配合物、所述方法制备得到的配合物中的至少一种。
可选地,所述纳米材料的制备方法,将配合物研磨后,通入(5%氢气+95%氮气),300℃加热3小时,还原得到纳米材料。
作为一种具体地实施方式,所述纳米材料的制备方法包括:将配合物研磨后,放于三角炉中,通入5%的氢气(95%为氮气)还原,设置温度为300℃,时间为3h,即得到所需要的纳米材料。
根据本申请的又一方面,提供一种催化剂,其特征在于,所述催化剂包括配合物还原得到的,瓜环包裹的纳米钯;所述配合物选自所述的配合物、所述方法制备得到的配合物中的至少一种。
可选地,所述催化剂的制备方法,包括:将所述配合物还原得到瓜环包裹的纳米钯材料;将所述瓜环包裹的纳米钯材料与卡铂碳混合,得到混合物i;将混合物i加入到含有异丙醇与水的混合溶液i中,然后加入nafion溶液,超声混合均匀,得到混合溶液ii,即所述催化剂;
其中,混合溶液i中异丙醇与水的体积比为1:1;混合溶液i与nafion溶液的体积比为300~200:1。
可选地,所述瓜环包裹的纳米钯材料与卡铂碳的质量比为1:1。
可选地,混合溶液i与nafion溶液的体积比为250:1。
作为一种具体地实施方式,所述催化剂的制备方法,包括以下步骤:
1、将配合物用氢气还原得到瓜环包裹的纳米钯材料;
2、将所述负载在瓜环上的纳米钯材料与卡铂碳放入同一个玻璃瓶,加入溶液异丙醇,水,nafion,超声;
3、将得到的分散有材料的液体,滴在碳纸上,作为电极,进行电化学性能测试。
作为一种具体的实施方式,所述催化剂的制备方法包括:
将还原得到的纳米钯材料与卡铂碳混合,放于异丙醇:水=1:1的混合溶液中,加入nafion溶液超声至混合均匀。即可得到二氧化碳还原电催化剂,即所述催化剂。
作为一种实施方式,所述材料是以氯化钯为前驱体,以十甲基五元瓜环为配体,制备了pdme10cb[5]配合物,用氢气将其还原后,得到瓜环包裹的纳米钯材料。
所述的催化剂的制备方法,包括如下步骤:
(1)首先合成十甲基五元瓜环作为超分子组装的配体;
(2)向氯化钯(0.17mmol)中加入去离子水(20ml),加入6m的盐酸水溶液1ml,形成氯化钯的透明黄色水溶液(溶液ⅰ);
(3)在磁力搅拌下将me10cb[5](100mg,0.1mmol)溶解在去离子水(20ml)中,形成无色透明水溶液(溶液ⅱ);
(4)将溶液ⅰ与ⅱ分别小心地转移到h管的每一侧。通过缓慢扩散在h管中,三天后获得棕色晶体;
(5)将得到的晶体研磨,在三角炉中选择不同的温度和时间进行氢化;性能最佳的条件是在300℃,3h;
(6)将氢化还原后的材料,与卡铂碳按照1:1的质量比混合,在异丙醇:水=1:1的混合溶液中超声至混合均匀。即得所述催化剂。
根据本申请的又一方面,提供一种所述的催化剂作为二氧化碳还原阴极催化剂的应用。
本申请中,me表示甲基;cb[n]表示瓜环类化合物,如cb[5]表示五元瓜环、cb[6]表示六元瓜环、me10cb[5]表示十甲基五元瓜环;me4cb[6]表示对称四甲基六元瓜环。
本申请能产生的有益效果包括:
(1)本申请中利用扩散法合成晶体,合成方法简单。生成的晶体结晶度高,在空气中稳定。
(2)利用5%的氢气还原晶体,还原速度缓慢,防止纳米颗粒因生成速度较快造成的团聚。
(3)在还原过程中,晶体中的氢键减弱了纳米钯的团聚,同时,瓜环的羰基由于略带负电性,通过静电作用结合在带正电的纳米钯的表面,减弱纳米颗粒的团聚。因此,得到的材料颗粒均一,活性位点充分暴露。
(4)本发明所制备的催化剂pd@me10cb[5]的选择性优异,尤其在较小的电位-0.6vvs.rhe(相对可逆氢电极)表现出高的一氧化碳选择性,接近90%。降低了催化剂的成本,同时提高了贵金属pd的利用率。
(5)本发明所制备的催化剂pd@me10cb[5]的稳定性较高,在一氧化碳选择性最高的电位(-0.6vvs.rhe)进行稳定性测试9h,电流效率保持为原来的91%。
(6)该催化剂为二氧化碳新型催化剂的研究提供了一定的经验及借鉴。
附图说明
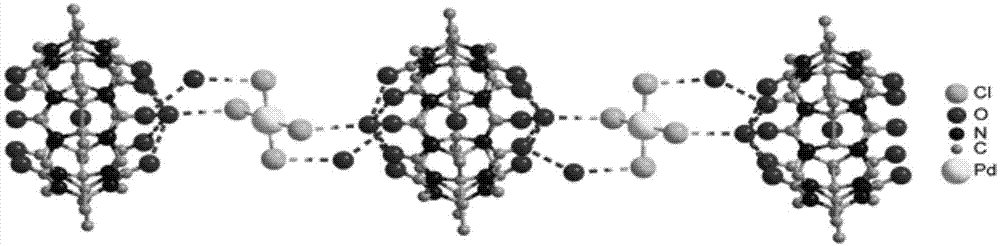
图1为实施例1制备的pdme10cb[5]的晶体结构图。
图2为实施例3制备的pd纳米材料的tem图,其中a为300℃,3h条件下氢化得到的pd纳米材料的tem图,b为pd纳米材料的高分辨率透射电镜图,插图为多晶pd的fft图。
图3为实施例5制备的pd@me10cb[5]催化剂的一氧化碳法拉第效率图。
图4为实施例5制备的pd@me10cb[5]催化剂的一氧化碳电流密度图。
图5为实施例5制备的pd@me10cb[5]催化剂在电位(-0.6vvs.rhe)时9h的稳定性测试。
具体实施方式
下面结合实施例详述本申请,但本申请并不局限于这些实施例。
如无特别说明,本申请的实施例中的原料和催化剂均通过商业途径购买,其中氯化钯购自国药公司;
十甲基五元瓜环me10cb[5]根据文献合成,合成原料中多聚甲醇和2,3-丁二酮均购自alfa公司。参考文献:lih,lüj,linj,etal.crystallinehybridsolidmaterialsofpalladiumanddecamethylcucurbit[5]urilasrecoverableprecatalystsforheckcross‐couplingreactions[j].chemistry-aeuropeanjournal,2013,19(46):15661-15668.
浓盐酸,乙醇和丙酮均购自上海国药。
本申请的实施例中分析方法如下:
电化学性能和电化学稳定性测试的仪器为:zahnerim6,型号在为im6,厂家为德国zahner(札纳)。
催化剂稳定性测试的仪器为zahnerim6,型号为im6,厂家为德国zahner(札纳)。
催化剂的形貌表征仪器为:场发射透射电子显微镜feitecnaig2f20,型号为feitecnaig2f20,厂家为美国fei公司。
实施例1晶体的生长方法
(1)向氯化钯(0.17mmol)中加入去离子水(20ml),6m的盐酸溶液1ml形成氯化钯的透明水溶液(溶液ⅰ)。在磁力搅拌下将me10cb[5](100mg,0.1mmol)溶解在去离子水(20ml)中(溶液ⅱ)。将溶液ⅰ与ⅱ分别小心地转移到h管的每一侧。在h管中,通过缓慢扩散(静置)三天,获得棕色晶体。
(2)将得到的晶体用去离子水洗涤3遍,在空气中自然干燥得到pdme10cb[5]晶体。记为样品1#,其中pd的含量为9.86wt%。
实施例2晶体结构解析
以上x–射线单晶衍射数据都是利用supernovaccd单晶衍射仪于100k下收集,对样品1#进行结构解析。衍射光源为石墨单色化的mo-kα射线
单晶测试结果如表1和表2所示。
表1
表2样品1#的原子坐标(不含氢原子)
样品1#的晶体结构(不含h)如图1所示。
由图1可以看出,样品1#中,游离水通过氢键结合在十甲基五元瓜环的两端,相邻的分子胶囊通过氢键和pdcl42-形成一维链状结构。水合氢离子起到平衡电荷的作用。
实施例3钯纳米材料的制备方法
将得到的晶体pdme10cb[5],放在石英管中,置于三角炉,通入5%的氢气还原(气氛为体积分数为5%的氢气,体积分数为95%的氮气)。设置三角炉温度为300℃,反应时间为3h。即可得到负载在瓜环上的纳米钯。命名为d-1#。
实施例4钯纳米材料的形貌表征
实施例3制备得到的还原的钯纳米材料样品d-1#进行形貌表征,样品d-1#的tem图如图2a和图2b所示。图2a显示,钯纳米材料呈均匀的颗粒状球形,粒径在2.2nm左右;图2b通过计算纳米钯的高分辨图表明晶格间距为0.22nm,相对应纳米钯的(111)面。插图为纳米钯的傅里叶变换图像,表明纳米钯的多晶结构,与pd(111)面取向一致。
实施例5钯纳米催化剂的制备方法
(1)将还原得到的纳米钯(d-1#)与相同质量的卡铂碳混合,分散在1ml超纯水和异丙醇的混合液中(体积比1:1),再加入40μl的萘酚nafion溶液,超声2h。即可得到掺杂卡铂碳的纳米钯催化剂。命名为d-2#。
实施例6电极制备
(1)工作电极:取240μl实施例5中的掺杂卡铂碳的纳米钯催化剂的混合液滴在碳纸表面(面积1*1cm2),干燥,即得;
(2)铂网为对电极,参比电极为ag/agcl。最后负载在电极表面的催化剂中金属钯的含量为120μg/cm2。
实施例7电化学活性测试
先在co2氛围下的0.5m碳酸氢钾溶液中进行循环伏安扫描,扫描速度为100mv·s-1,扫描范围为-1.2~0vvs.rhe(相对可逆氢电极),扫描10圈,该步骤的作用是对催化剂表面进行清洁并起到一定的活化作用。然后进行恒电压测试,气相色谱(gc)检测产物,1hnmr检测液相产物,dmso作为内标,以上扫描范围选取参比电极为ag/agcl电极,pt网为对电极。
图3至图5为实施例5所提供的催化剂,即样品d-2#的电化学性能测试图。从图中可以看出本申请所合成的催化剂拥有较高的选择性,稳定性和催化活性。
在测试过程中,只检测到氢气和一氧化碳。图3为样品d-2#pd@me10cb[5]催化剂产物的法拉第效率图,可以看到,该材料在较低的电位(-0.6vvs.rhe)下,表现出高的一氧化碳法拉第效率,接近90%,随着过电位的升高,氢气的竞争反应,导致一氧化碳的法拉第效率降低。而较低的过电位正是我们所需要的。
图4为样品d-2#pd@me10cb[5]催化剂的产物电流密度图。可以看出,随着过电位的升高,一氧化碳和氢气的电流密度增加,在电位(-1.1vvs.rhe)时,一氧化碳的电流密度是氢气电流密度的2倍。
实施例8电化学稳定性测试
图5为样品d-2#pd@me10cb[5]催化剂在电位(-0.6vvs.rhe)时9h的稳定性测试,图5显示,反应进行9h后,相对电流密度还能保持为原来的91%,表现出较高的稳定性。
以上所述,仅是本申请的几个实施例,并非对本申请做任何形式的限制,虽然本申请以较佳实施例揭示如上,然而并非用以限制本申请,任何熟悉本专业的技术人员,在不脱离本申请技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
- 还没有人留言评论。精彩留言会获得点赞!