一种S掺杂的金属有机框架材料的合成方法与流程
本发明涉及一种非金属元素掺杂的金属有机框架材料放入合成,尤其涉及一种s掺杂的金属有机框架材料的合成方法,主要作为超级电容器赝电容器材料,属于金属有机合成技术领域和超级电容器的储能材料领域。
背景技术
金属有机框架材料(metalorganicframeworks,mofs)是近年来发展迅速的一种有机、无机化学结合而成的一种多孔材料,是继氧化石墨烯和碳纳米管之外的一类新型多孔材料。mofs是一种有机配体与金属离子或金属离子簇通过配位键形成的二维或三维的骨架结构,有序的孔道结构以及较大的比表面积在荧光、吸附、分离、储气和催化等诸多领域有巨大的应用价值。mofs材料独特的金属离子或离子簇中心,提供有成有效的活性位点,改善了电化学电荷的传递,因此mofs材料可作为一种超级电容器的储能材料备受学者关注。
mofs材料作为超级电容器赝电容器材料,疏松有序的孔道结构有利于电解液的渗透,金属离子或离子簇的存在是发生氧化反应的本质所在,同样体现了储能机制。非金属原子修饰的mofs材料,增多了mofs材料的活性位点,使过快放电得到有效改善,因此,非金属元素对低储能mofs的改性可进一步提高材料的储能性能。
技术实现要素:
本发明的目的是提供一种s掺杂的金属有机框架材料的合成方法,以改善金属有机框架材料的储能性能。
一、s掺杂的金属有机框架材料的合成的合成
本发明s掺杂的金属有机框架材料s@ni-mof的合成,是以将mofs前驱体分散于水中形成ni-mof的悬浮液,并加热到80~85℃,然后将硫脲溶液逐滴加入到ni-mof的悬浮液中,持续搅拌2~3h,冷却后转移至反应釜中,于170~180℃反应4~5h,反应结束后,抽滤,洗涤,干燥,研磨,获得s掺杂的金属有机框架材料s@ni-mof。前驱体ni-mof和硫脲的质量为1:2~1:2.5。
前驱体ni-mof的制备:将均苯三甲酸与4,4,-联吡啶溶解在n,n-二甲基甲酰胺中得溶液
所述洗涤为:先用dmf洗去未反应的均苯三甲酸、4,4,-联吡啶以及盐,再用蒸馏水洗去dmf及未洗干净的盐,然后用乙醇洗涤。
所述干燥为于60~70℃真空干燥20~24小时。
二、s@ni-mof的表征
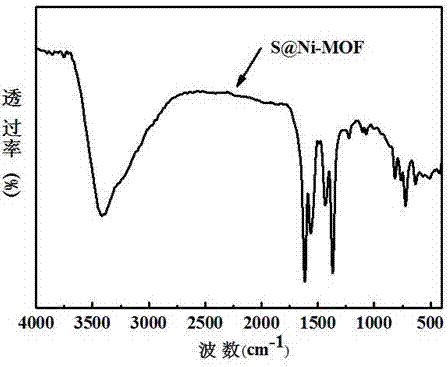
1、ft-ir分析
图1为本发明合成的s@ni-mof材料的ft-ir图。3416cm-1处的峰是未参与配位的-cooh中-oh伸缩振动峰,位于1619cm-1、1065cm-1和815cm-1处的吸收峰是c-n键的伸缩振动峰,位于1369cm-1处的吸收峰是c=o键的振动峰。
2、扫描电镜分析
图2为本发明合成的s@ni-mof材料的扫描电镜图。扫描电子显微镜照片表明,s@ni-mof是由片片堆积的均一多孔花状材料,这种疏松的结构裸露出更多的活性位点,有利于电荷的转移。
3、热重分析
图3为本发明合成的s@ni-mof材料的热分析图。热分析图表明,本发明合成的s@ni-mof材料热稳定性非常好,从室温~800℃,失重率仅为17.6%。300℃之前的失重是溶剂分子的挥发以及未反应有机配体的升华,300℃~600℃的失重归结为合成过程中小分子聚合物的分解,600℃之后的失重归结为结构的坍塌。
4、x射线粉末衍射分析
图4为本发明合成的s@ni-mof材料的x射线粉末衍射图。x射线粉末衍射图表明,s@ni-mof材料具有较好的结晶度。较好的结晶度说明材料合成过程中生长去向单一,电化学性能测试过程中并无其他生长取向的材料对其影响,保证电化学性能测试材料的唯一性。
5、eds面扫元素分布分析
图5为本发明合成的s@ni-mof材料的eds元素分析图。其中,(a)为s@ni-mof材料中富含所有元素分布图,(b)为s@ni-mof材料中c元素的分布图,(c)为s@ni-mof材料中n元素的分布图,(d)为s@ni-mof材料中o元素的分布图,(e)为s@ni-mof材料中ni元素的分布图,(f)为s@ni-mof材料中s元素的分布图。从图看出s元素均匀的分布在ni-mof材料的表面,说明s元素掺杂到前驱体ni-mof材料中。
6、前驱体和s@ni-mof相同电流密度的比电容比较
图6为本发明合成的s@ni-mof与前驱体ni-mof材料相同电流密度下恒电流充放电曲线图。从图6中可以看到在相同电流密度下,掺杂s元素的s@ni-mof具有较长的放电时间和较快宽的电位窗口,而前驱体ni-mof表现出短的放电时间和窄的电位窗口。因此,s@ni-mof材料的电化学性能优于前驱体ni-mof材料。
7、循环伏安测试分析
图7为本发明合成的s@ni-mof材料的循环伏安图。在0~0.6v的电位窗口下,扫速由5mvs-1增加到100mvs-1的cv曲线。图中存在显著的氧化还原峰,表现出典型的赝电容性能;而且随着扫速的增大,出现极化现象,氧化还原峰向电势两端移动,同时扫速的增大,电势差变化基本一致,说明具有良好的可逆性。良好的可逆性,有利于s@ni-mof材料的多次循环使用。
8、恒电流充放电测试分析
图8为本发明合成的s@ni-mof材料恒电流充放电曲线图。从图8中可以发现,充放电曲线并呈现“小房子”,显示出明显的法拉第反应。此外,随着充电流密度的逐渐增大,其比容值并未大幅度的衰减,其优异的倍率性能可看出该材料良好的可逆性。
9、交流阻抗测试分析
图9为本发明合成的s@ni-mof材料煅烧后的交流阻抗图。在高频区,呈现半圆状,且半径较小,说明s@ni-mof材料自身内阻较小;在中低频区,代表warburg扩散阻抗,该水系电解液离子要进入到电极的内部中相对较小的孔隙中去,电解液离子的运动就属于扩散动力学控制,其特点是斜率越大,阻抗值越小。即斜率越大越有利于离子在活性物质内部的扩散迁移;斜率越小,阻碍作用越明显。
综上所述,本发明采用水热法合成了s掺杂的多孔金属有机框架材料,表现出良好的热稳定性以及电化学性能,说明前驱体材料ni-mof材料经s元素修饰,有效改善了mofs放电过程及储能性能,提高了ni-mof材料的热稳定性,作为超级电容器电极具有很好的应用前景。
附图说明
图1为本发明合成的s@ni-mof材料的红外光谱图;
图2为本发明合成的s@ni-mof材料的扫描电镜图;
图3为本发明合成的s@ni-mof材料的热分析图;
图4为本发明合成的s@ni-mof材料的x射线粉末衍射图;
图5为本发明合成的s@ni-mof材料的eds面扫元素分析图;
图6为本发明合成的s@ni-mof与前驱体ni-mof材料的恒电流充放电的放电曲线图;
图7为本发明合成的s@ni-mof材料的循环伏安图;
图8为本发明合成的s@ni-mof材料恒电流充放电的放电曲线图;
图9为本发明合成的s@ni-mof材料的交流阻抗图。
具体实施方式
下面通过具体实施例对本发明s@ni-mof材料的合成和性能作进一步说明。
实施例1
前驱体ni-mof的合成:将0.021g(1.0mmol)均苯三甲酸、0.019g(1.2mol)4,4,-联吡啶溶解在7mln,n-二甲基甲酰胺(dmf)中为溶液
s@ni-mof的合成:取0.6~0.65g前驱体ni-mof,分散到8~10ml水中,得到前驱体的悬浮液;并将悬浮液加热至80~85℃;取硫脲0.8~0.88g,溶解于6~8ml水中,制得硫脲水溶液;然后逐滴滴加至ni-mof前驱体悬浮液中,持续搅拌2~3h;冷却后转移至反应釜中,于170~180℃反应4~5h;反应结束后,抽滤,洗涤,60~70℃真空干燥20~24小时,研磨,获得非金属掺杂的s@ni-mof。
电化学性能测试:将上述制备的s@ni-mof材料在1ag-1,2ag-1,4ag-1,5ag-1,6ag-1,8ag-1,10ag-1,15ag-1和20ag-1的电流密度下,比电容值分别为1453.54fg-1,1297.50fg-1,1160.83fg-1,1111.46fg-1,1070.13fg-1,1008.83fg-1,953.75fg-1,860.00fg-1,774.17fg-1。说明s@ni-mof材料具有较大的比电容值。
实施例2
前驱体ni-mof的合成:同实施例1;
s@ni-mof的合成:取1.2~1.3g前驱体ni-mof,分散到8~10ml水中,得到前驱体的悬浮液;并将悬浮液加热至80~85℃;取硫脲0.8~0.88g,溶解于6~8ml水中,制得硫脲水溶液;然后逐滴滴加至ni-mof前驱体悬浮液中,持续搅拌2~3h;冷却后转移至反应釜中,于170~180℃反应4~5h;反应结束后,抽滤,洗涤,60~70℃真空干燥20~24小时,研磨,获得非金属掺杂的s@ni-mof-2。
电化学性能测试:将上述制备的s@ni-mof-2材料在1ag-1的电流密度下,比电容值分别为1089.25fg-1。说明s@ni-mof-2材料在前驱体ni-mof的增加,导致部分前驱体未被s掺杂,二者的混合拉低了比容值。
实施例3
前驱体ni-mof的合成:同实施例1;
s@ni-mof的合成:取0.6~0.65g前驱体ni-mof,分散到8~10ml水中,得到前驱体的悬浮液;并将悬浮液加热至80~85℃;取硫脲0.4~0.44g,溶解于6~8ml水中,制得硫脲水溶液;然后逐滴滴加至ni-mof前驱体悬浮液中,持续搅拌2~3h;冷却后转移至反应釜中,于170~180℃反应4~5h;反应结束后,抽滤,洗涤,60~70℃真空干燥20~24小时,研磨,获得非金属掺杂的s@ni-mof-3。
电化学性能测试:将上述制备的s@ni-mof-2材料在1ag-1的电流密度下,比电容值分别为1238.46.fg-1。说明s@ni-mof-3材料在合成中分散剂尿素量的减少,导致前驱体ni-mof分散不完全,少量的前驱体ni-mof未被s元素掺杂,影响比容值的降低。
实施例4
前驱体ni-mof的合成:同实施例1;
s@ni-mof的合成:取0.6~0.65g前驱体ni-mof,分散到8~10ml水中,得到前驱体的悬浮液;并将悬浮液加热至80~85℃;取硫脲0.8~0.88g,溶解于6~8ml水中,制得硫脲水溶液;然后逐滴滴加至ni-mof前驱体悬浮液中,持续搅拌2~3h;冷却后转移至反应釜中,于130~150℃反应4~5h;反应结束后,抽滤,洗涤,60~70℃真空干燥20~24小时,研磨,获得非金属掺杂的s@ni-mof-3。
电化学性能测试:将上述制备的s@ni-mof-3材料在1ag-1的电流密度下,比电容值分别为562.39fg-1。说明s@ni-mof-3材料在水热温度较低时,反应不充分,大量的前驱体ni-mof存在,导致比容值的降低。
实施例5
前驱体ni-mof的合成:同实施例1;
s@ni-mof的合成:取0.6~0.65g前驱体ni-mof,分散到8~10ml水中,得到前驱体的悬浮液;并将悬浮液加热至80~85℃;取硫脲0.8~0.88g,溶解于6~8ml水中,制得硫脲水溶液;然后逐滴滴加至ni-mof前驱体悬浮液中,持续搅拌2~3h;冷却后转移至反应釜中,于170~180℃反应1~2h;反应结束后,抽滤,洗涤,60~70℃真空干燥20~24小时,研磨,获得非金属掺杂的s@ni-mof-4。
电化学性能测试:将上述制备的s@ni-mof-4材料在1ag-1的电流密度下,比电容值分别为729.63fg-1。说明s@ni-mof-4材料在水热时间较短时,反应不充分,部分前驱体ni-mof与s元素复合,二者混合导致比容值的降低。
- 还没有人留言评论。精彩留言会获得点赞!