一种L-草胺膦关键中间体的合成新方法与流程
本发明涉及除草剂有机中间体的合成技术领域,尤其涉及一种l-草胺膦关键中间体的合成新方法。
背景技术:
草胺膦(glufosinate-ammonium)是一种广谱、低毒、非选择性除草剂,由hoechest公司在上世纪80年代开发生产。草胺膦是全球第二大转基因作物耐受除草剂,应用前景十分广阔。草铵膦具有两种对映异构体,但只有l-构型具有除草活性,4-(羟基甲基膦酰基)-2-羰基丁酸是化学法合成l-构型草铵膦的关键中间体。近年来,化学法和生物法合成l-草胺膦的方法被相继开发出来,但毫无例外的均需要使用关键中间体4-(羟基甲基膦酰基)-2-羰基丁酸。
专利us06936444公开了processforthepreparationofl-phosphinothricinebyenzymatictransaminationwithaspartate(利用天冬氨酸盐通过酶转胺化制备l-膦酯的方法),该方法的制备过程为:在生物法中,4-(羟基甲基膦酰基)-2-羰基丁酸利用转氨酶作用将羰基转化为氨基而选择性得到l-草胺膦;hoechest公司在1991(j.org.chem,vol.56,no.51991)最先报道了4-(羟基甲基膦酰基)-2-羰基丁酸的化学合成法:利用甲基亚膦酸单乙酯与丙烯酸乙酯在乙醇钠的作用下发生迈克尔加成先制得3-(乙氧基甲基膦酰基)-丙酸乙酯,然后在零下50度与草酸二乙酯在乙醇钠作用下发生克莱森酯缩合,再用盐酸水解脱羧后制得4-(羟基甲基膦酰基)-2-羰基丁酸。安徽国星生物化学有限公司在专利cn106008596中公开了一种草胺膦中间体4-(羟基甲基磷酰基)-2-羰基丁酸的制备方法,江苏七洲绿色化工股份有限公司在专利cn103665032中公开了一种草铵膦的制备方法,这两种方法均是采用环膦酸酐与草酸二乙酯或氰化钠制备4-(羟基甲基膦酰基)-2-羰基丁酸。
现有的制备方法中,hoechest法需要深冷到零下50℃,且需要使用价格昂贵的乙醇钠,反应总收率低;环膦酸酐法所使用的环膦酸酐制备不易,成本高,且纯化困难,容易导致其酸度值较高,在克莱森缩合中导致醇钠被额外消耗造成反应体系复杂,总收率低,而使用酸度值较高的环磷酸酐与氰化钠反应时会造成巨大的安全隐患,且氰化钠本身为剧毒,应尽量避免使用。
技术实现要素:
有鉴于此,本发明提供了一种l-草胺膦关键中间体的合成新方法,该方法操作简便,成本低、工艺环境友好,对生产设备无特殊要求,特别适用于工业化生产,利用该方法制得的产物经结晶析出,hplc纯度可达98%以上。
本发明提供一种l-草胺膦关键中间体的合成新方法,包括以下步骤:
s101,在第一溶剂或无溶剂条件下,将式ii结构的化合物与式iii结构的化合物混合,反应后蒸馏,得到式iv结构的化合物:
s102,在第二溶剂的条件下,将式iv结构的化合物与有机酸混合,反应后蒸馏,得到式v结构的化合物:
s103,在质子性溶剂的条件下,将式v结构的化合物、海因与弱碱混合,反应后加入第一酸溶液调节ph值,然后过滤、洗涤、干燥,得到式vi结构的化合物:
s104,向式vi结构的化合物中加入强碱溶液,反应后得到式vii结构的化合物:
s105,向式vii结构的化合物中加入第二酸溶液调节ph值,然后蒸干水得到固体物,利用第三溶剂溶解固体物,过滤、冷却,加入第四溶剂,再次过滤,洗涤、干燥,即得到式i结构的化合物:
式ii、式iv、式v、式vi中,r1为c1-c4的烷基;式iii中,x为氯、溴或碘。
进一步地,步骤s101中,所述式ii结构的化合物与式iii结构的化合物混合后在80-130℃下反应5-12h。
进一步地,步骤s101中,所述式ii结构的化合物与式iii结构的化合物的摩尔比为1:1-1.5,所述第一溶剂选用乙腈、1,4-二氧六环、1,2-二氯丙烷、甲苯或二甲苯中的任一种。
进一步地,步骤s102中,所述式iv结构的化合物与有机酸混合后在30-90℃下反应2-12h,所述式iv结构的化合物与有机酸的摩尔比为1:0.1-0.5。
进一步地,步骤s102中,所述第二溶剂选择二氯甲烷、甲苯、乙腈、1,4-二氧六环或四氢呋喃中的任一种,所述有机酸选择为三氟甲磺酸、草酸、对甲苯磺酸或甲基磺酸中的任一种。
进一步地,步骤s103中,所述式v结构的化合物、海因与弱碱混合后在50-100℃下反应8-12h,所述式v结构的化合物、海因与弱碱的摩尔比为1:1:0.8-1。
进一步地,步骤s103中,所述质子性溶剂选择水、乙醇、甲醇或异丙醇中的任一种,所述弱碱选择碳酸氢钠、碳酸氢钾、乙醇胺或三乙胺中的任一种,所述第一酸溶液为盐酸。
进一步地,步骤s104中,向式vi结构的化合物中加入强碱溶液后,在50-100℃下反应1-12h,所述式vi结构的化合物与强碱溶液的摩尔比为1:2-3。
进一步地,步骤s104中,所述强碱溶液选择氢氧化钠溶液。
进一步地,步骤s105中,所述第二酸溶液选择盐酸或硫酸的任一种,所述第三溶剂选择丙酮,所述第四溶剂选择甲基异丁基酮。
本发明提供的技术方案带来的有益效果是:本发明提供的l-草胺膦关键中间体的合成新方法先以式ii结构的化合物与式iii结构的化合物通过arbuzov重排反应生成式iv结构的化合物,再用有机酸对其解缩得到式v结构的化合物,式v结构的化合物与海因环在催化剂作用下进行缩合反应得到式vi结构的化合物,式vi结构的化合物经过碱水解得到式vii结构的化合物,最后对式vii结构的化合物调酸后结晶得到式i结构的化合物,即4-(羟基甲基膦酰基)-2-羰基丁酸;相比现有的合成路线,本发明提供的合成路线均为常规反应,原料易得,成本低,反应条件温和,收率高且环境友好,具有很大的工业化前景,利用本发明提供的方法制得的4-(羟基甲基膦酰基)-2-羰基丁酸经结晶析出,hplc纯度可达98%以上。
附图说明
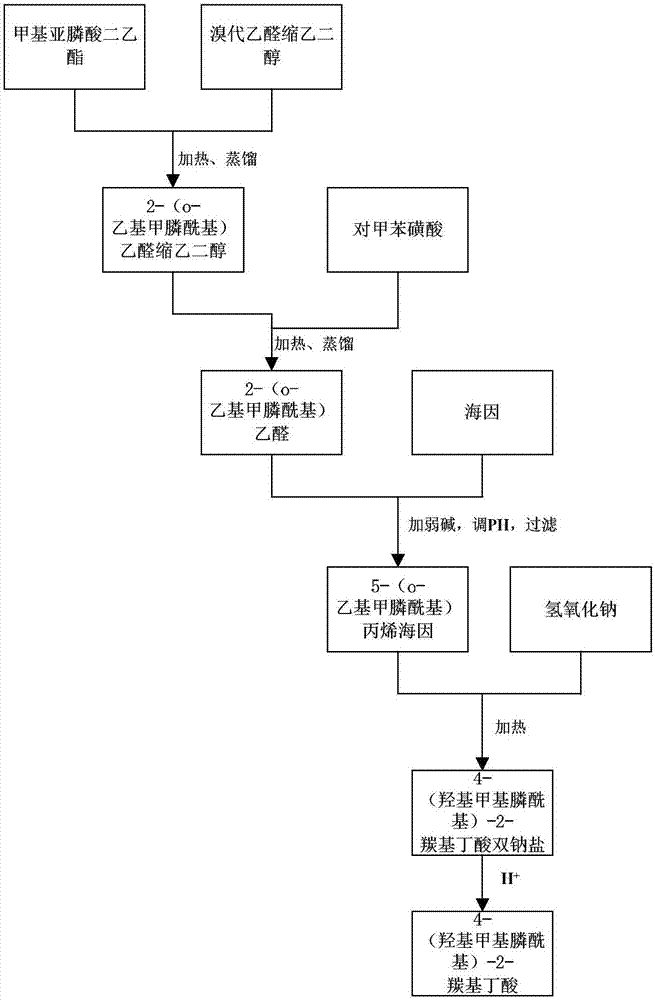
图1是本发明实施例2一种l-草胺膦关键中间体的合成新方法的流程示意图。
具体实施方式
为使本发明的目的、技术方案和优点更加清楚,下面将结合附图对本发明实施方式作进一步地描述。
本发明的实施例提供了一种l-草胺膦关键中间体的合成新方法,包括以下步骤:
步骤s101,在第一溶剂或无溶剂条件下,将式ii结构的化合物与式iii结构的化合物混合,在80-130℃下反应5-12h,反应后减压蒸馏去除低沸物,得到式iv结构的化合物;
式ii结构的化合物的结构式为:
式iii结构的化合物的结构式为:
式iv结构的化合物的结构式为:
步骤s101中,式ii结构的化合物与式iii结构的化合物的摩尔比为1:1-1.5,第一溶剂选用乙腈、1,4-二氧六环、1,2-二氯丙烷、甲苯或二甲苯中的任一种。
步骤s102,在第二溶剂的条件下,将式iv结构的化合物与有机酸混合,在30-90℃下反应2-12h,反应后减压蒸馏,得到式v结构的化合物;
式v结构的化合物的结构式为:
步骤s102中,式iv结构的化合物与有机酸的摩尔比为1:0.1-0.5,第二溶剂选择二氯甲烷、甲苯、乙腈、1,4-二氧六环或四氢呋喃中的任一种,有机酸选择为三氟甲磺酸、草酸、对甲苯磺酸或甲基磺酸中的任一种。
步骤s103,将式v结构的化合物与海因(乙内酰脲,cas号:461-72-3)混合,然后加入弱碱,再加入质子性溶剂,在50-100℃下反应8-12h,反应后加入第一酸溶液调节ph值至4,然后过滤、洗涤、干燥,得到式vi结构的化合物;
式vi结构的化合物的结构式为:
步骤s103中,式v结构的化合物、海因与弱碱的摩尔比为1:1:0.8-1,质子性溶剂选择水、乙醇、甲醇或异丙醇中的任一种,弱碱选择碳酸氢钠、碳酸氢钾、乙醇胺或三乙胺中的任一种,第一酸溶液为盐酸。
步骤s104,向式vi结构的化合物中加入强碱溶液,在50-100℃下反应1-12h,反应后得到式vii结构的化合物;
式vii结构的化合物的结构式为:
步骤s104中,式vi结构的化合物与强碱溶液的摩尔比为1:2-3,强碱溶液选择20%的氢氧化钠溶液。
步骤s105,向式vii结构的化合物的水溶液中加入第二酸溶液调节ph值至1-2,然后蒸干水得到呈黄色粘稠物状的固体物,利用第三溶剂溶解固体物,过滤,得到的滤液冷却至0℃左右,缓慢搅拌,然后缓慢滴加第四溶剂直至反应体系开始变浑浊,在此温度下析晶5-12h后再次过滤,利用第四溶剂洗涤再次过滤得到的滤饼,真空干燥,即得到式i结构的化合物,即l-草胺膦的关键中间体4-(羟基甲基膦酰基)-2-羰基丁酸;
式i结构的化合物的结构式为:
步骤s105中,第二酸溶液选择盐酸或硫酸的任一种,第三溶剂选择丙酮,第四溶剂选择甲基异丁基酮。
式ii、式iv、式v、式vi中,r1为c1-c4的烷基,式ii中的两个r1可以相同,也可以不同;式iii中,x为氯、溴或碘。
上述l-草胺膦关键中间体的合成路线为:
下面结合实施例对本发明提供的一种l-草胺膦关键中间体的合成新方法进行详细说明。
实施例1:
在500ml的四口烧瓶中加入甲基亚膦酸二甲酯108g(1mol)、溴代乙醛缩乙二醇167g(1mol),氮气保护下加热,在120℃下回流反应5h(gc检测反应终点),冷却至室温,将反应液减压蒸馏,10mmhg下收集120℃馏分,得到2-(o-甲基甲膦酰基)乙醛缩乙二醇164g,产率91.1%;
在250ml的三口烧瓶中加入2-(o-甲基甲膦酰基)乙醛缩乙二醇36g(0.2mol)、三氟甲磺酸3g(0.02mol)、乙腈100ml,在30℃下反应5h(gc检测反应终点),将反应液减压蒸馏,15mmhg下收集110℃馏分,得到呈无色液体的2-(o-甲基甲膦酰基)乙醛26g,产率95.6%,2-(o-甲基甲膦酰基)乙醛的分析结果为:1hnmr(400mhz,cdcl3,δppm):1.28(t,j=7.2hz,3h),1.53(d,j=14.5hz,3h),3.12(d,j=17.6hz,2h),4.08(t,j=7.4hz,2h),9.70(s,1h),31pnmr(400mhz,cdcl3,δppm):45.18;
在250ml的三口烧瓶中加入海因15g(0.15mol)和碳酸氢钾12g(0.12mol),再加入100ml水,油浴缓慢升温到70℃,同时在1h内匀速滴加2-(o-甲基甲膦酰基)乙醛20.4g(0.15mol),滴加过程中固体逐渐溶解至澄清,搅拌回流,保持90℃反应8h,反应液呈淡黄色,停止反应后冷却至室温,有少量固体析出,用6n盐酸调节ph至4,析出大量固体,抽滤,水洗,干燥,得到白色的5-(o-甲基甲膦酰基)丙烯海因30g,产率91.7%;
在100ml的三口烧瓶中加入5-(o-甲基甲膦酰基)丙烯海因22g(0.1mol)与20%的氢氧化钠溶液50ml,65℃下搅拌回流7h,得到4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液;
在4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液加入浓盐酸调节ph值至1.5,将水蒸干得黄色粘稠状物质,加入少量丙酮溶解,过滤滤除无机盐,得到的丙酮溶液在0℃左右进行缓慢搅拌,再缓慢加入甲基异丁基酮直到体系变浑浊,持续搅拌8h后过滤,利用甲基异丁基酮洗涤过滤得到的滤饼,真空干燥,得到白色粉末状的4-(羟基甲基膦酰基)-2-羰基丁酸13.3g,产率73.9%。
实施例2:
图1是本发明实施例2制备4-(羟基甲基膦酰基)-2-羰基丁酸的流程示意图。
在1l的四口烧瓶中加入甲基亚膦酸二乙酯136g(1mol)、溴代乙醛缩乙二醇167g(1mol)和甲苯400ml,氮气保护下加热,在110℃下回流反应5h(gc检测反应终点),冷却至室温,将反应液减压蒸馏,10mmhg下收集120℃馏分,得到2-(o-乙基甲膦酰基)乙醛缩乙二醇182g,产率93.8%;
在250ml的三口烧瓶中加入2-(o-乙基甲膦酰基)乙醛缩乙二醇38.8g(0.2mol)、对甲苯磺酸6.88g(0.04mol)、甲苯100ml,在50℃下反应6h(gc检测反应终点),将反应液减压蒸馏,12mmhg下收集108℃馏分,得到呈无色液体的2-(o-乙基甲膦酰基)乙醛29g,产率96.7%;
在250ml的三口烧瓶中加入海因15g(0.15mol)和三乙胺15.2g(0.15mol),再加入100ml乙醇,油浴缓慢升温到60℃,同时在1h内匀速滴加2-(o-乙基甲膦酰基)乙醛22.5g(0.15mol),滴加过程中固体逐渐溶解至澄清,搅拌回流,保持75℃反应10h,反应液呈淡黄色,反应结束后将溶剂蒸除,冷却至室温,用6n盐酸调节体系ph至4,析出大量固体,抽滤,水洗,干燥,得白色的5-(o-乙基甲膦酰基)丙烯海因32g,产率92%;
在100ml的三口烧瓶中加入5-(o-乙基甲膦酰基)丙烯海因23.2g(0.1mol)与20%的氢氧化钠溶液50ml,85℃下搅拌回流6h,得到4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液;
在4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液加入浓盐酸调节ph值至1-2,将水蒸干得黄色粘稠状物质,加入少量丙酮溶解,过滤滤除无机盐,得到的丙酮溶液在0℃左右进行缓慢搅拌,再缓慢加入甲基异丁基酮直到体系变浑浊,持续搅拌8h后过滤,利用甲基异丁基酮洗涤过滤得到的滤饼,真空干燥,得到白色粉末状的4-(羟基甲基膦酰基)-2-羰基丁酸12g,产率66.7%。
实施例2制得的4-(羟基甲基膦酰基)-2-羰基丁酸的分析检测结果如下:
1hnmr(400mhz,dmso-d6)酮式:δ10.93(s,2h,oh),3.00(dt,j=10.5,7.6hz,2h,pch2ch2co),1.86-1.76(m,2h,pch2ch2co),1.31(dd,j=22.7,14.0hz,3h,pch3);烯醇式:δ10.93(s,2h,oh),5.52(dt,1h,pch2ch=c),2.56(dd,2h,pch2ch=),1.29(d,3h,pch3)。
31pnmr(400mhz,dmso-d6,δppm)酮式:46.81,烯醇式44.82。hplc纯度98.2%。
hplc条件:色谱柱ultimatexb-c18,5μm,4.6x250mm;柱温:30℃;检测波长:205nm,进样量6μl,目标化合物保留时间:10.0min。
实施例3:
在500ml的四口烧瓶中加入甲基亚膦酸二正丙酯164g(1mol),氯代乙醛缩乙二醇123g(1mol),氮气保护下加热,在120℃下回流反应9h(gc检测反应终点),冷却至室温,将反应液减压蒸馏,10mmhg下收集125馏分,得到2-(o-正丙基甲膦酰基)乙醛缩乙二醇176g,产率84.6%;
在250ml的三口烧瓶中加入2-(o-正丙基甲膦酰基)乙醛缩乙二醇41.6g(0.2mol)、草酸7.56g(0.06mol)、四氢呋喃100ml,在70℃下反应8h(gc检测反应终点),将反应液减压蒸馏,12mmhg下收集120℃馏分,得到呈无色液体的2-(o-正丙基甲膦酰基)乙醛28g,产率85.4%;
在250ml的三口烧瓶中加入海因15g(0.15mol)和碳酸氢钠11.8g(0.14mol),再加入100ml水,油浴缓慢升温到65℃,同时在1h内匀速滴加2-(o-正丙基甲膦酰基)乙醛24.8g(0.15mol),滴加过程中固体逐渐溶解至澄清,搅拌回流,保持85℃反应8.5h,反应液呈淡黄色,停止反应后冷却至室温,有少量固体析出,用6n盐酸调节ph至4,析出大量固体,抽滤,水洗,干燥,得到白色的5-(o-丙基甲膦酰基)丙烯海因35.6g,产率95.7%;
在100ml的三口烧瓶中加入5-(o-丙基甲膦酰基)丙烯海因24.8g(0.1mol)与20%的氢氧化钠溶液50ml,95℃下搅拌回流7h,得到4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液;
在4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液加入浓盐酸调节ph值至1-2,将水蒸干得黄色粘稠状物质,加入少量丙酮溶解,过滤滤除无机盐,得到的丙酮溶液在0℃左右进行缓慢搅拌,再缓慢加入甲基异丁基酮直到体系变浑浊,持续搅拌9h后过滤,利用甲基异丁基酮洗涤过滤得到的滤饼,真空干燥,得到白色粉末状的4-(羟基甲基膦酰基)-2-羰基丁酸12.7g,产率70.5%。
实施例4:
在500ml的四口烧瓶中加入甲基亚膦酸二正丁酯192g(1mol)、氯代乙醛缩乙二醇123g(1mol)、乙腈400ml,氮气保护下加热,在80℃下回流反应12h(gc检测反应终点),冷却至室温,将反应液减压蒸馏,8mmhg下收集130℃馏分,得到2-(o-正丁基甲膦酰基)乙醛缩乙二醇175g,产率78.8%;
在250ml的三口烧瓶中加入2-(o-正丁基甲膦酰基)乙醛缩乙二醇44.4g(0.2mol)、甲基磺酸7.68g(0.08mol)、1,4-二氧六环100ml,在90℃下反应12h(gc检测反应终点),将反应液减压蒸馏,10mmhg下收集125℃馏分,得到呈无色液体的2-(o-正丁基甲膦酰基)乙醛29g,产率81.5%;
在250ml的三口烧瓶中加入海因15g(0.15mol)和乙醇胺7.9g(0.13mol),再加入100ml水,油浴缓慢升温到90℃,同时在1h内匀速滴加2-(o-正丁基甲膦酰基)乙醛26.6g(0.15mol),滴加过程中固体逐渐溶解至澄清,搅拌回流,保持95℃反应6h,反应液呈淡黄色,停止反应后冷却至室温,有少量固体析出,用6n盐酸调节ph至4,析出大量固体,抽滤,水洗,干燥,得到白色的5-(o-丁基甲膦酰基)丙烯海因36.0g,产率92.3%;
在100ml的三口烧瓶中加入5-(o-丁基甲膦酰基)丙烯海因26g(0.1mol)与20%的氢氧化钠溶液50ml,95℃下搅拌回流4h,得到4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液;
在4-(羟基甲基膦酰基)-2-羰基丁酸双钠盐的水溶液加入浓盐酸调节ph值至1.8,将水蒸干得黄色粘稠状物质,加入少量丙酮溶解,过滤滤除无机盐,得到的丙酮溶液在0℃左右进行缓慢搅拌,再缓慢加入甲基异丁基酮直到体系变浑浊,持续搅拌6h后过滤,利用甲基异丁基酮洗涤过滤得到的滤饼,真空干燥,得到白色粉末状的4-(羟基甲基膦酰基)-2-羰基丁酸14.1g,产率78.4%。
本发明提供的l-草胺膦关键中间体4-(羟基甲基膦酰基)-2-羰基丁酸的合成新方法先以式ii结构的化合物与式iii结构的化合物通过arbuzov重排反应生成式iv结构的化合物,再用有机酸对其解缩得到式v结构的化合物,式v结构的化合物与海因环在催化剂作用下进行缩合反应得到式vi结构的化合物,式vi结构的化合物经过碱水解得到式vii结构的化合物,最后对式vii结构的化合物调酸后结晶得到式i结构的化合物,即4-(羟基甲基膦酰基)-2-羰基丁酸;相比现有的合成路线,本发明提供的合成路线均为常规反应,原料易得,成本低,反应条件温和,收率高且环境友好,具有很大的工业化前景,利用本发明提供的方法制得的4-(羟基甲基膦酰基)-2-羰基丁酸经结晶析出,hplc纯度可达98%以上。
在不冲突的情况下,本文中上述实施例及实施例中的特征可以相互结合。
以上所述仅为本发明的较佳实施例,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!