多相丙烯共聚物的制作方法
本公开涉及具有低含量的低聚物并且特别适合于汽车元件特别是用于汽车内饰元件的多相丙烯共聚物。
背景技术:
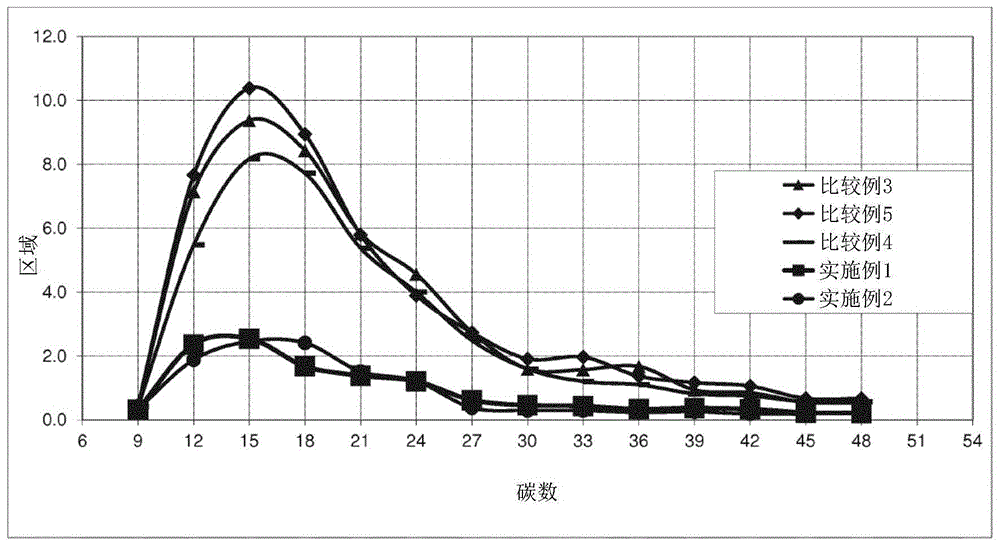
:聚丙烯是许多应用的选择材料。例如,聚丙烯组合物用于汽车内饰的制品中。一般而言,所述组合物是良好的可加工性并且可以单独定制。用于汽车内饰的丙烯组合物的问题可能是恶臭和起雾。这些挥发性化合物的存在是造成新车内饰经典气味的原因。wo2012/049204涉及具有低含量低聚物的多相丙烯共聚物,其是通过含有两个外部电子给体的特定催化剂体系获得的。技术实现要素:申请人发现,通过使用包含铋和至少一种具有通式的外部电子给体化合物的特定催化剂体系,可以进一步减少挥发性化合物(低聚物)的量:(r1)asi(or2)b其中r1和r2独立地选自具有1-8个碳原子的烷基中,可选地含有杂原子,并且a是0或1并且a+b=4。本公开的目的是聚丙烯组合物,其包含:a)50wt%至90wt%,具有在25℃下高于90%的不溶于二甲苯的部分的丙烯均聚物,并且具有mfrl(根据iso1133,条件l,即230℃和2.16kg载荷的熔体流动速率)0.5至200g/10min;b)10重量%至50重量%;具有30.0重量%至70.0重量%的乙烯衍生单元的丙烯和乙烯的共聚物;组分a)和b)的量之和为100;该组合物具有:i)在25℃下可溶于二甲苯的部分的特性粘度为2.2至4.0dl/g;ii)mfrl(根据iso1133,条件l,即230℃和2.16kg载荷的熔体流动速率)为0.5至100g/10min;iii)二甲苯可溶部分为10重量%至50重量%;所述聚丙烯组合物通过两步聚合工艺获得,其中:步骤a)在包含以下反应之间的反应产物的催化剂的存在下,使丙烯聚合以获得组分a):a)包含ti、mg、cl和内部电子给体化合物的固体催化剂组分,其特征在于相对于所述固体催化剂组分的总重量,其包含0.1至50wt%的bi;b)烷基铝化合物,以及c)具有通式的外部电子给体化合物:(r1)asi(or2)b其中r1和r2独立地选自具有1-8个碳原子的烷基中,可选地含有杂原子,并且a是0或1并且a+b=4。步骤b)在步骤a)的聚合产物的存在下,将丙烯和乙烯聚合以获得组分b)。附图说明图1表示实施例1和2以及比较例3-5的tds分析值的图。图2表示实施例6和比较例7的tds分析值的图。具体实施方式本公开的目的是聚丙烯组合物,其包含:a)50重量%至90重量%;优选60重量%至85重量%;更优选67重量%至82重量%,具有在25℃下高于90%、优选高于95%、更优选高于97%的不溶于二甲苯的部分的丙烯均聚物;和mfrl(根据iso1133,条件l,即230℃和2.16kg载荷的熔体流动速率)为0.5至200g/10min,优选为50至150g/10min,更优选为80至140g/10min;b)10重量%至50重量%;优选15重量%至40重量%;更优选18重量%至33重量%;丙烯和乙烯的共聚物,其具有30.0重量%至70.0重量%,优选35.0重量%至60.0重量%,更优选40.0重量%至58.0重量%,甚至更优选45重量%至55%的乙烯衍生单位;组分a)和b)的量之和为100;该组合物具有:i)在25℃下可溶于二甲苯的部分的特性粘度为2.2至4.0dl/g;优选在2.5和4.0dl/g之间,更优选在2.6和3.5dl/g之间;ii)mfrl(根据iso1133,条件l,即230℃和2.16kg载荷的熔体流动速率)为0.5至100g/10min,优选为8至70g/10min,更优选为10至60g/10min;iii)二甲苯可溶部分为10重量%至50重量%;优选12重量%至35重量%;所述聚丙烯组合物通过两步聚合工艺获得,其中:步骤a)在包含以下反应之间的反应产物的催化剂的存在下,使丙烯聚合以获得组分a):a)包含ti、mg、cl和内部电子给体化合物的固体催化剂组分,其特征在于相对于所述固体催化剂组分的总重量,其包含0.1至50wt%的bi;b)烷基铝化合物,以及c)具有通式的外部电子给体化合物:(r1)asi(or2)b其中r1和r2独立地选自具有1-8个碳原子的烷基中,可选地含有杂原子,并且a是0或1并且a+b=4。步骤b)在步骤a)的聚合产物的存在下,将丙烯和乙烯聚合以获得组分b)。根据以上定义,显而易见的是,术语“共聚物”是指仅包含两种共聚单体的聚合物。根据本公开的聚丙烯组合物优选用于汽车内饰元件。对于汽车内饰元件,是指汽车的所有内饰零件,尤其是门把手、门袋、装饰和包裹架子、风道、加热器/空调单元壳体、用于仪器板的电枢、中央控制台、地毯。用于本公开的内饰汽车元件的聚丙烯组合物的低聚物含量非常低,特别是在前反应器聚合物中测得的低聚物含量低于2000ppm;优选低于1500ppm。此外,本公开的用于汽车内饰元件的聚丙烯组合物在23℃、0℃和-20℃时具有改善的伊佐德值,并保持了良好的拉伸模量值。优选地,拉伸模量在800mpa至1600mpa之间,优选地在850mpa至1500mpa之间;低聚物的极低值以及机械性能使得本公开的组合物适合用于汽车内饰,从而减少气味和挥发性化合物的排放。因此,本公开的另一个目的是包含本公开的聚丙烯组合物的汽车内饰。本文公开的聚烯烃组合物是通过以下工艺制备的,该工艺包括在第一步中将丙烯均聚,然后在第二步中将丙烯与乙烯均聚,这两种步骤均在包含以下反应产物的催化剂的存在下进行:a)一种包含ti、mg、cl和至少一种电子给体化合物的固体催化剂组分,其特征在于,相对于所述固体催化剂组分的总重量,其包含0.1至50重量%的bi;b)烷基铝化合物,以及c)具有通式的外部电子给体化合物:(r1)asi(or2)b其中r1和r2独立地选自具有1-8个碳原子的烷基中,可选地含有杂原子,并且a是0或1并且a+b=4。优选地,在催化剂组分中,bi的含量的范围为0.5至40重量%,更优选为1至35重量%,尤其是2至25重量%,在非常具体的实施方案中为2至20重量%。固体组分的颗粒具有基本球形的形态,并且平均直径在5-150μm之间,优选20-100μm,更优选30-90μm。作为具有基本球形形态的颗粒,是指长轴和短轴之间的比率等于或小于1.5,优选小于1.3。通常,mg的量的范围优选为8至30重量%,更优选为10至25重量%。通常,ti的量的范围为0.5-5重量%,更优选0.7-3重量%。优选的内部电子给体化合物选自可选地取代的芳族多元羧酸的烷基和芳基酯,例如苯甲酸和邻苯二甲酸的酯。此类酯的具体实例为邻苯二甲酸正丁酯、邻苯二甲酸二异丁酯、邻苯二甲酸二正辛酯、苯甲酸乙酯和对乙氧基苯甲酸乙酯。mg/ti摩尔比优选等于或高于13,优选在14-40的范围内,更优选在15至40。相应地,mg/给体摩尔比优选大于16更优选大于17,通常在18至50的范围。bi原子优选衍生自一种或多种不具有bi-碳键的bi化合物。特别地,bi化合物可以选自卤化bi、碳酸bi、乙酸bi、硝酸bi、氧化bi、硫酸bi、硫化bi。bi的化合价为3+的化合物是优选的。在卤化bi中,优选三氯化bi和三溴化bi。最优选的bi化合物是bicl3。固体催化剂组分的制备可以根据几种方法进行。根据一种方法,可以通过使式为ti(or)q-yxy的钛化合物(其中q为钛的化合价,y为1至q之间的数字,优选为ticl4)与衍生自式mgcl2·proh的加合物的氯化镁(其中p是0.1至6,优选2至3.5的数,r是具有1-18个碳原子的烃基)反应来制备固体催化剂组分。通过在搅拌条件下在加合物的熔融温度(100-130℃)下操作,将醇和氯化镁混合,可以适当地以球形制备加合物。然后,将加合物与与加合物不混溶的惰性烃混合,从而形成乳液,其被快速淬灭,从而导致球形颗粒形式的加合物固化。根据该程序制备的球形加合物的实例描述于usp4,399,054和usp4,469,648中。如此获得的加合物可以与ti化合物直接反应,或者可以预先进行热控制的脱醇(80-130℃),从而获得其中醇的摩尔数通常小于3,优选在0.1和2.5之间的加合物。与ti化合物的反应可以通过将加合物(脱醇的或类似的物质)悬浮在冷的ticl4(通常为0℃)中进行;将该混合物加热至80-130℃,并在该温度下保持0.5-2小时。ticl4处理可以进行一次或多次。电子给体化合物可以在用ticl4处理期间以期望的比例加入。有几种方法可以在催化剂制备中添加一种或多种bi化合物。根据优选的选择,将bi化合物在其制备过程中直接掺入mgcl2·proh加合物中。特别地,可以通过将其与mgcl2和醇混合在一起而在加合物制备的初始阶段添加bi化合物。或者,可以在乳化步骤之前将其添加到熔融加合物中。相对于加合物中每摩尔mg,bi的引入量为0.1至1摩尔。直接掺入mgcl2·proh加合物中的优选bi化合物是卤化bi,尤其是bicl3。烷基-al化合物(ii)优选选自三烷基铝化合物,例如三乙基铝、三异丁基铝、三正丁基铝、三正己基铝、三正辛基铝。还可能使用烷基铝卤化物、烷基铝氢化物或倍半氯化烷基铝,例如alet2cl和al2et3cl3,可能与上述三烷基铝混合。al/ti比大于1,通常介于50和2000之间。外部电子给体化合物(c)是具有通式的硅化合物(r1)asi(or2)b(ii)其中r1和r2独立地选自具有1-8个碳原子的烷基,可选地包含杂原子,其中a为0或1且a+b=4。第一组优选的式(ii)的硅化合物是a为1,b为3且r1和r2分别选自具有2-6个,优选2-4个碳原子的烷基,特别优选异丁基三乙氧基硅烷(ibtes)。另一组优选的式(ii)的硅化合物是其中a为0,b为4并且r2独立地选自具有2-6个,优选2-4个碳原子的烷基,特别优选四乙氧基硅烷。外电子给体化合物(c)的用量应使有机铝化合物与所述外电子给体化合物(iii)之间的摩尔比为0.1至200,优选为1至100,更优选为3至50。聚合工艺在本领域中是已知的,例如在一个或多个流化或机械搅拌床反应器中的气相操作,使用惰性烃溶剂作为稀释剂的淤浆聚合或使用液体单体的本体聚合(例如丙烯)作为反应介质。本公开的聚丙烯组合物还可以在两个或更多个阶段中通过聚合工艺获得,其中在第一阶段中获得组分a),然后在第二阶段中在组分a)存在下获得组分b)。每个阶段可以是气相的,可以在一个或多个流化或机械搅拌的床反应器中进行,淤浆相可以使用惰性烃溶剂作为稀释剂,也可以使用液态单体(例如丙烯)作为反应介质进行本体聚合。聚合通常在20至120℃,优选40至80℃的温度下进行。当聚合在气相中进行时,操作压力通常为0.5至5mpa,优选为1至4mpa。在本体聚合中,操作压力通常在1和8mpa之间,优选在1.5和5mpa之间。氢通常用作分子量调节剂。用于本公开的组合物还可包含本领域通常使用的添加剂,例如抗氧化剂,光稳定剂,热稳定剂,成核剂,着色剂和填充剂。根据本公开的汽车内饰元件可以通过本领域中通常已知的技术,例如通过注射成型,热成型等,从上述的聚丙烯组合物开始获得。给出以下实施例以说明而不是限制本公开。实施例mg,ti的确定固体催化剂组分中mg和ti含量的确定是通过“i.c.p光谱仪arlaccuris”上的电感耦合等离子体发射光谱法进行的。通过在“fluxy”铂坩埚中分析称重0.1÷0.3克催化剂和2克偏硼酸锂/四硼酸锂1/1混合物来制备样品。加入几滴ki溶液后,将坩埚插入特殊设备“claissefluxy”中进行完全燃烧。用5%v/vhno3溶液收集残留物,然后bi的确定固体催化剂组分中bi含量的确定已通过“i.c.p光谱仪arlaccuris”上的电感耦合等离子体发射光谱法进行。通过在200cm3容量瓶中分析称重0.1÷0.3克催化剂来制备样品。缓慢加入约10毫升65%v/vhno3溶液,约50cm3的蒸馏水后,样品经过4÷6小时的消化。然后用去离子水将容量瓶稀释至刻度。通过icp在以下波长下直接分析所得溶液:铋,223.06nm。内部给体含量的确定通过气相色谱法确定固体催化化合物中内部给体的含量。将固体组分溶解在丙酮中,加入内标,并在气相色谱仪中分析有机相的样品,以确定起始催化剂化合物上存在的给体的量。x.i.和x.s.的确定将2.5g聚合物和250cm3邻二甲苯引入装有冰箱和磁力搅拌器的玻璃烧瓶中。在30分钟内将温度从室温升至溶剂的沸点(135℃)。然后将如此获得的澄清溶液保持回流并再搅拌30分钟。然后将密闭的烧瓶在25℃的恒温水浴中保持30分钟,以使样品的不溶物(xi)发生结晶。如此形成的固体在快速滤纸上过滤。将100cm3的过滤后的液体倒入预先称重的铝制容器中,该容器在氮气流下在加热板上加热,以通过蒸发除去溶剂。然后将容器在真空中保持在80℃的烘箱中干燥,然后在获得恒重后称重。因此,人们计算出在25℃下可溶于和不溶于二甲苯的聚合物的重量百分比。特性粘度(i.v.)将样品在135℃下溶于四氢萘中,然后倒入毛细管粘度计中。粘度计管(ubbelohde型)被圆柱形玻璃外套包围;此设置允许使用循环的恒温液体进行温度控制。弯液面的下行通道通过光电装置定时。在上部灯前面的弯液面通道启动了具有石英晶体振荡器的计数器。当弯液面经过下部灯时,弯液面使计数器停止,并记录流出时间:通过huggins方程将其转换为特性粘度值(huggins,m.l.,j.am.chem.soc.,1942,64,2716),前提是在相同的实验条件下(相同的粘度计和相同的温度)已知纯溶剂的流动时间。一种单一的聚合物溶液用于确定[η]。熔体流动速率(mil)根据iso1133(230℃,2.16kg)确定聚合物的熔体流动速率mil。丙烯/乙烯共聚物的13cnmr13cnmr光谱是在装有低温探针的brukerav-600光谱仪上获得的,该光谱仪在120℃下以160.91mhz的傅里叶变换模式工作。sββ碳的峰(根据“通过13cnmr测量的乙烯-丙烯橡胶中的单体序列分布。3.反应概率模式的使用”c.j.carman,r.a.harringtonandc.e.wilkes,大分子,1977,10,536)用作29.9ppm的内部参考。将样品在120℃下以8%wt/v的浓度溶解在1,1,2,2-四氯乙烷-d2中。每个光谱都是通过90°脉冲,脉冲和cpd之间的15秒延迟来消除1h-13c耦合而获得的。使用9000hz的频谱窗口,将512个瞬态存储在32k数据点中。光谱的分配,三重态分布的评估和组合物是根据kakugo(“用δ-三氯化钛-二乙基氯化铝制备的乙烯-丙烯共聚物中的单体序列分布的碳13nmr确定”m.kakugo,y.naito,k.mizunuma和t.miyatake,大分子,1982,15,1150)使用以下方程式进行的:ppp=100tββ/sppe=100tβδ/sepe=100tδδ/spep=100sββ/spee=100sβδ/seee=100(0.25sγδ+0.5sδδ)/ss=tββ+tβδ+tδδ+sββ+sβδ+0.25sγδ+0.5sδδ使用以下方程评估乙烯含量的摩尔百分比:e%mol=100*[pep+pee+eee]乙烯含量的重量百分比使用以下方程评估:其中p%mol是丙烯含量的摩尔百分比,而mwe和mwp分别是乙烯和丙烯的分子量。根据carman(c.j.carman,r.a.harrington和c.e.wilkes,大分子,1977;10,536)计算反应比r1r2的乘积为:丙烯序列的立构规整度是根据pppmmtββ(28.90-29.65ppm)和整个tββ(29.80-28.37ppm)之比计算为mm含量。组分b)的乙烯含量已经通过使用下式由乙烯的总含量计算:乙烯(b重量%)=乙烯(总的重量%/(b量的重量%)/100)。拉伸模量拉伸模量是根据iso527-2和iso1873-2测量的。低聚物含量通过溶剂萃取确定低聚物含量包括在25℃的超声波浴中,用10ml的亚甲基二氯化物(ch2cl2)处理5g的聚丙烯样品4小时。将1μl提取液注入毛细管柱,并使用fid分析,无需任何过滤。为了定量估计低聚物含量,已应用基于外标法的校准。特别地,已经使用了一系列烃(c12-c22-c28-c40)。tds分析将0.10至0.15g之间的量的聚合物装入敞开的玻璃管(177mm长,6mm外径,4mm内径)中,然后将其插入tds3热解吸器(gerstel,mülheimanderruhr,德国)安装在配备有ms-5973n质谱检测器(电子倍增器1500,ms范围从29到800)的agilentgc-6890n(安捷伦科技,比勒利卡,ma,美国)色谱仪上。该仪器在烘箱中装有ptv进样器和hp-5msui部件n°19091s-436ui(安捷伦科技)色谱柱(60m长,0.25mm内径,0.10微米膜厚)。热解吸器的温度以60℃/min的速率从40℃升高到280℃,然后在280℃下保持15分钟,同时使用液氮将注射器在-50℃下冷冻以提取低聚物,添加剂及其副产品。该工艺发生在超纯氦中,以提供无氧气氛。然后将解吸的物质注入色谱柱中。在洗脱期间,烤箱的温度从40℃(5分钟等温步骤)到320℃,以6c°/min的速率变化,耗时101.67分钟。载气是超纯氦气,恒定流速为0.7ml/min。色谱图是在全扫描模式下获取的总离子流(tic)。通过安捷伦化学工作站软件进行详细说明。实施例1和2制备球形加合物的程序根据wo98/44009的比较例5中所述的方法制备微球状mgcl2·pc2h5oh加合物,区别在于在进料油之前已添加了粉末形式的bicl3,相对于镁的量为3mol%。固体催化剂组分的制备程序在室温,氮气氛下,向配备有机械搅拌器,冷却器和温度计的500ml圆底烧瓶中引入300mlticl4。冷却至0℃后,在搅拌下,将邻苯二甲酸二异丁酯和9.0g的球形加合物(如上所述制备)依次加入烧瓶中。内部给体的进料量应满足mg/给体摩尔比为8。将温度升至100℃并保持2小时。之后,停止搅拌,使固体产物沉降,并在100℃下虹吸除去上清液。去除上清液后,再添加新鲜的ticl4,以再次达到初始液体体积。然后将混合物在120℃加热并在该温度下保持1小时。再次停止搅拌,使固体沉降并且虹吸除去上清液。在低至60℃的温度梯度下用无水己烷洗涤该固体六次,并在室温下洗涤一次。然后将获得的固体在真空下干燥并分析。预聚合处理如表1所示,在将其引入聚合反应器之前,将如上所述的固体催化剂组分与三乙基铝(teal)和异丁基三乙氧基硅烷(ibtes)或四乙氧基硅烷(teos)接触。聚合聚合运行在连续两个反应器中以连续模式进行,该反应器配备有将产物从一个反应器转移到紧邻的反应器中的装置。第一个反应器是液相循环反应器,第二个反应器是流化床气相反应器。在液相循环反应器中制备丙烯均聚物,而在来自第一阶段的丙烯均聚物存在下,在气相反应器中制备丙烯乙烯共聚物。氢气用作分子量调节剂。通过气相色谱法连续分析气相(丙烯、乙烯和氢)。在运行结束时,将粉末排出并在氮气流下干燥。表1报道了与反应器中生产的聚合物有关的主要聚合条件和分析数据。表2报道了聚合物的性能。比较例3-5固体催化剂组分的制备比较例3-5的催化剂通过使用与实施例1的催化剂相同的方法生产,但是使用表1中报道的甲基环己基二甲氧基硅烷(c给体)或二环戊基二甲氧基硅烷(d给体)。预聚合处理已根据实施例1的程序进行了预聚合处理聚合通过使用与实施例1相同的程序进行聚合。表1列出了与反应器中生产的聚合物有关的主要聚合条件和分析数据。表2列出了聚合物的性能。表1c2-=乙烯;c3-=丙烯;h2=氢表2*计算从表2得出的结果是,根据本公开获得的丙烯组合物的低聚物含量明显低于通过使用不同的外部给体获得的比较例之一。实施例6制备球形加合物的程序根据wo98/44009的比较例5中所述的方法制备微球状mgcl2·pc2h5oh加合物,区别在于在进料油之前已添加了粉末形式的bicl3,相对于镁的量为3mol%。固体催化剂组分的制备程序在室温,氮气氛下,向配备有机械搅拌器,冷却器和温度计的500ml圆底烧瓶中引入300mlticl4。冷却至0℃后,在搅拌下加入9.0g的球形加合物(如上所述制备),然后将3,3-二丙基戊二酸二乙酯依次加入烧瓶中。内部给体的加入量应满足mg/给体摩尔比为13。将温度升至100℃并保持2小时。之后,停止搅拌,使固体产物沉降,并在100℃下虹吸除去上清液。虹吸后,加入新鲜的ticl4和一定量的9,9-双(甲氧基甲基)芴,以使mg/二醚摩尔比为13。然后将混合物在120℃加热并在搅拌下在该温度下保持1小时。再次停止搅拌,使固体沉降并且虹吸除去上清液。在低至60℃的温度梯度下用无水己烷洗涤该固体六次,并在室温下洗涤一次。然后将获得的固体在真空下干燥并分析。预聚合处理在将其引入聚合反应器之前,将上述固体催化剂组分与表3中的三乙基铝(teal)和异丁基-三乙氧基硅烷(ibtes)接触。聚合聚合运行按照实施例1进行。聚合物的性能列于表4。比较例7固体催化剂组分的制备通过使用与实施例6的催化剂相同的方法制备比较例7的催化剂。预聚合处理如表3所示,在将其引入聚合反应器之前,使上述固体催化剂组分与三乙基铝(teal)接触。聚合通过使用与实施例5相同的程序进行聚合。关于在反应器中生产的聚合物的主要聚合条件和分析数据记录在表3中。聚合物的性能记录在表4中。表3表4实施例5比较例6外部给体类型ibtesc组分a)均聚物含量%8685mfrg/10'130140xs%1.71.4组分b)共聚物含量*%1415组合物的性能乙烯含量重量%6.77.0二甲苯可溶部分%12,814,1二甲苯可溶部分的特性粘度dl/g2,742,37mfrg/10'64,067,8拉伸模量mpa14101480低聚物ppm14701660当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1