应用全基因组捕获转座子间区段序列对受微生物污染的人类DNA样本进行基因组序列分析的制作方法
电子提交的参考序列表
序列表的正式版本,是以ascii格式通过epct电子方式提交,文档大小2.5千字节(kilobytes),称为“phgl1002.wogenesequencelisting”,于2018年5月18日创建,并与说明书一并提交。所述ascii格式文档中的序列表属该说明书的一部分,引用时全部内容将并入说明书。
本发明大致涉及一种序列和结构变异的分析方法,可应用在被微生物dna污染的人类dna样品上。具体地说,本发明利用基因组中众多alu、mir和sva逆转录转座子插入位点,以多核苷酸链式反应(pcr)及基于alu-、mir-、sva-共有序列设计的pcr引物,扩增转座子间(ite)的基因组区段,并通过‘almivascan’测序平台进行大规模并行测序(mps)对扩增子进行序列分析。由于没有任何相关报道指出细菌和病毒dna中存在alu、mir和sva逆转录转座子,采用所述almivascan测序方法,共存微生物dna对人类基因组分析的干扰将会非常小。这开启了almivascan的应用,使被细菌和病毒污染的人类分泌物和排泄物dna的序列分析变得可行。
背景技术:
虽然有多种方法平台能有效率地分析人类基因组序列,包括全基因组测序(“wgs”)和全外显子测序(“wes”),但是对dna样本的需求是不能被其他物种dna污染。因此,诸如痰液中常带有肺和上呼吸道组织dna,包括肺癌和上呼吸道癌的dna,及粪便中常含有胃肠组织dna,包括胃肠道癌dna;但因为这些痰液或粪便dna往往受到肺部和上呼吸道的细菌和病毒及胃肠道的生物群落污染,使dna分析变得困难。由于alu逆转录转座子仅在灵长类动物dna中发现,mir逆转录转座子仅在哺乳动物中发现,及sva逆转录转座子仅在人科动物中发现,那使用基于alu-、mir-和sva-共有序列设计的pcr引物扩增人类基因组dna时,即使当中存在细菌和病毒dna,仍会获得具人类dna特异性的扩增子,序列分析也不会受到干扰。因此,本发明的目的是要开发和测试所述方法的实用性,用于分析受微生物污染的人类分泌物和排泄物如痰液和粪便中的人类dna。痰液中含有来自肺部和上呼吸道的癌症和癌前细胞,而粪便中含有来自胃肠的癌细胞。所以,若可以对这些样本中的人类dna进行分析,将加速早期癌症检测,及监测治疗后癌细胞存活和/或变异的情况。
转座子是单细胞基因组中一段可移动的dna序列。转座的发生能改变细胞的基因组,造成表型上的显著突变。人类最常见的转座子形式是逆转录转座子,包括alu序列,mir序列和sva序列,是人类基因组中的重复序列家族。通常,alu元件长约280个碱基对,mir元件长约145个碱基对,均为重复性dna元件类别,且同属短散在元件(“sines”),而sva元件长约1000个碱基对。在人类基因组中,大约有1,080,000个alu重复序列、595,000个mir重复元件和3,700个sva重复元件。
本文所用术语“alu元件”包含一短段dna,具有限制性内切核酸酶alu(藤黄节杆菌)的识别序列。在灵长类动物基因组中存在着大量不同种类的alu元件。事实上,alu元件是人类基因组中最丰富的转座元件。它们源于小胞浆7slrna,是信号识别颗粒的一个组成部分。alu序列的典型结构是5'-a部分-a5taca6-b部分-polya尾-3'(seqidno:1)。由于a部分和b部分的核苷酸序列相似,其结构揭示了现代人类alu元件是在1亿年前由两种不同的古化石单体融合而成。alu家族中不同成员间的poly-a尾长度参差不一。首被发现的alu元件亚家族为aluj和alus,其它alu亚家族成员也很快被发现。而其中alus的其一亚亚家族因包含活跃alu元件,最终被命名为aluy。
本文所用术语“mir元件”包含久远的重复序列家族,广泛发生于胎盘哺乳动物、有袋动物和单孔目动物中,并有着序列差异,这显示它们为丰富的全哺乳动物的散在重复序列,或简称mirs。mir元件的典型结构是由一段trna源性区域与一无相关序列融合组成,而该trna源性区域包含rna聚合酶iii启动子共有序列,并与人类gln-trna-cug基因相似。
本文所用术语“vntr”代表可变数目串联重复序列,尽管不是所有串联重复结构域中的重复拷贝数均属多态性,但也包含微卫星和小卫星。本文所用术语“sva元件”涵盖由sine-vntr-alu组成的非自主人科动物逆转录转座子家族,由于svas正处于活跃状态,因此它们可以通过多种机制,包括突变、外显子改组、选择性剪接和通过产生差异性甲基化区域,导致基因组变异,甚至能改变人类性状。
表型是指任何受基因型影响的可观察参数,包括rna和蛋白质类分子。纵使大多数由遗传物质编码合成的分子和结构在生物体上是不可见的,但它们是可观察的(通过表型如细胞形态等),因此也属表型的一部分。人类血型就是一个典型例子,决定血型的基因型,其表型在细胞水平表达。因此,人类中的基因组序列和结构变异可应用于分析人类受试者之间的基因型及表型差异。在同一个体内,细胞也可能发生体细胞突变,导致基因型及表型改变;如导致癌症的恶性突变。
大规模并行测序技术(mps)彻底改变了遗传学的进程,它能通过一次运行产生千兆碱基序列信息,大大降低了wgs的成本,并已被广泛用于性状关联和疾病关联的研究。然而,其主要缺点为需要较大的dna量。在大多数情况下,甚至3微克的基因组dna仍未能完全达到全基因组测序的严格要求。人类分泌物和排泄物中所含的人类总dna量通常是有限的,要获取达微克量需求的dna样品就更加困难。
有鉴于此,须要采用新方法以求降低所需dna量和测序成本,产生高质相应序列数据。聚合酶链式反应技术(“pcr”)允许用者采集有限量的起始基因组dna,并从基因组不同区域进行扩增,获取足够dna量用于序列分析。然而,由于要从基因组不同区域扩增大量序列,需要使用很多昂贵的pcr引物,所以本发明开发了一种能选择性地扩增逆转录转座子间ite区段序列的方法,仅需使用少量特定设计的pcr引物,如基于alu元件共有序列设计的pcr引物。
美国专利号5,773,649,标题为“用于检测癌细胞突变体表型的dna标记物和诊断癌细胞的方法”,发明者为sinnett等人,于1998年6月30日发布,称之为“sinnett649专利”。sinnett649专利使用了配对alu间pcr(inter-alupcr)引物进行扩增,扩增片段与对应不稳定易感性基因座的探针杂交,并在聚丙烯酰胺凝胶上进行电泳分级,以确定肿瘤和无肿瘤dna之间是否存在变异条带。这种inter-alupcr系统,仅使用配对pcr引物,而pcr扩增子在凝胶电泳中呈条带状模式。
美国专利号7,537,889,标题为“alu元件于人类dna量化检测中的应用”,发明者为sinha等人,于2009年5月26日发布,称之为“sinha889专利”。sinha889专利使用了配对inter-alupcr引物对dna样本进行扩增,检测样本中人类dna的存在及数量,而当中可能存在非人类dna。检测方法是基于新近融入到人类基因组中的多拷贝alu元件,这些元件于大部分于非人类灵长动物和其他哺乳动物中均不存在。在sinha889专利中,这种alu间(inter-alu)pcr方法,是一种仅使用配对引物的pcr系统,而pcr扩增子在凝胶电泳中呈条带状模式。
美国专利申请号2015/0225722a1,标题为“针对构成非编码rna的异染色质的选择性靶向方法”,发明者是ozsolak,申请日为2015年4月29日。该方法是使用寡核苷酸来调节由非编码rna调控的基因异染色质状态。在一些实施方案中,此类基因包含三重重复区域或其它包含alu元件和mir元件的重复区域,但并没有使用基于alu-、mir-或sva-共有序列设计的pcr引物。
sinnett649专利和sinha889专利中所描述的发明,是利用传统pcr配合以alu-共有序列设计的配对引物进行扩增,获得于凝胶电泳后呈条带状的pcr扩增子。在多项科学研究中(kassandbatzer1995;walkeretal2003;krajinovicetal2012),基于alu序列的pcr被应用在法医案件检验工作,根据人类特异性扩增子条带模式,对含有其他物种dna的人类dna进行鉴定和定量。然而,在另一项mei、ding、xue等人所做的研究中(2011),便应用了alu间pcr对人类基因组dna的alu间序列进行大范围扩增,通过mps分析,产生具代表性的基因组序列扫描,被称为aluscan平台。为了提高alu间pcr扩增子序列的多样性,该平台采用了多条基于alu元件共有序列设计的pcr引物,引物分为头向(h-型,h-type)和尾向(t-型,t-type)。h-型引物是往alu逆转录转座子的头方向外延伸,而t-型引物则往alu逆转录转座子的尾方向外延伸。这些h-型和t-型引物能从亚微克dna样品中产出大量dna扩增子,经溴化乙锭染色后的凝胶电泳图呈弥散状而不是离散条带。通过mps对这些扩增子进行测序,可获取众多来自人类基因组不同位置的dna序列,经进一步分析,鉴定与人类性状的可能性关联。这种aluscan方法的重要性在于仅需少数基于alu序列设计的pcr引物及亚微克量dna模板,便能经扩增子捕获大量基因组序列片段进行mps分析。kumar、yang、xue等人(2015)所进行的研究,显示了aluscan方法的应用,有关研究并揭示癌症中有着大量间质性的杂合性丢失突变的发生。
aluscan平台的应用已通过对源自人类基因组不同区域的alu间序列进行扩增和测序被证实,而ng、hu、xue等人(2016)的研究更进一步发现人类基因组序列可依据基因组特征分为三种类型的序列区域,就是:451%的基因组序列位于富集基因、cpg岛、调控元件、甲基化位点和alu元件的基因区内,另31.1%位于富集增强子、保守indel位点和mir元件的近端区内,最后23.8%是在富含at碱基的远端区内,而基因区和近端区在功能上比远端区更重要。由于alu元件插入较多发生在基因区内:诸如aluj的基因区/近端区插入比率为2.83、alus为2.82、aluy为2.04,aluscan中所使用的基于alu共有序列设计的pcr引物,将有利于捕获基因区域内的alu间序列。为了均衡基因和近端两个区域中序列和结构变异的代表情况,在设计扩增基因组序列的pcr引物时,不仅采用alu元件共有序列,还需另一种富集于近端区段的转座子共有序列。人类基因组中存在大量mir元件,且主要富集于近端区域,其基因区/近端区插入比率为0.68。因此,使用基于alu-和mir-共有序列设计的引物进行pcr扩增,产生的alu间、mir间和alu-mir间扩增子,将能有效覆盖基因和近端两个序列区域。另外,要是想扩大覆盖范围,除了alu-和mir-序列基pcr引物外,更可增加使用以其它逆转录转座子序列设计的pcr引物进行序列捕获。就如sva元件,仅在人科动物的基因组中找到,属最年轻的转座子,主要存在于基因区域,并担当着重要的调控角色(quinnandbubb,2014;gianfrancesco,bubbandquinn,2017)。所以sva相邻序列会具有与人类性状相关的潜在基因组变异,这促使除了使用alu-和mir-序列基引物外,使用sva-序列基引物亦成为对ite-pcr引物的重要来源。
因此,采用alu共有序列基的h-型和t-型引物来进行alu间pcr,仅需使用亚微克量dna,便可获得大量alu间扩增子作测序分析之用。同样地,以相同的步骤,分别以mir共有序列基引物获得大量mir间扩增子及sva共有序列基引物获得大量sva间扩增子,并一拼使用alu共有序列基引物、mir共有序列基引物和/或sva共有序列基引物进行扩增,以获取大量alu间、mir间、sva间、alu-mir间、alu-sva间和mir-sva间扩增子进行测序分析。由于来自人类分泌物及排泄物的dna样品通常都受细菌和病毒dna污染,而干扰人类基因组测序的常规操作,所以能否以上述ite-pcr方法,使用alu-,mir-和/或sva序列基引物从受细菌和/或病毒dna污染的人类dna样本中成功扩增人类基因组序列进行mps分析,乃是关键所在。
技术实现要素:
通过转座子间聚合酶链式反应(ite-pcr),获取一系列高产量及高多样性的扩增子。以亚微克量人类dna为模板,于退火时,通过多条alu、mir和sva序列基的h-型和t-型引物分别从各逆转录转座子的头部和尾部延伸;随后使用大规模并行测序(mps)对这些ite-pcr扩增子进行序列分析,生成‘almivascan’dna序列扫描,当中包含alu间、mir间、sva间、alu-mir间、alu-sva间和mir-sva间区段序列。本发明利用pcr扩增alu、mir及sva逆转录转座子邻近区域序列,发现所捕获的序列当中明显富集基因组的序列和结构变异,例如单核苷酸多态性、微卫星、种系拷贝数变异和体细胞拷贝数变异,显著性达p<0.01。由于个人和细胞性状与基因组变异互相关联,‘almivascan’中的dna序列所提供的依据,有助分析基因组序列和结构变异与个人或细胞性状之间的关联,包括非医学性状、患病状态、疾病易感性和药物反应。本发明可应用于受细菌和病毒dna污染的人类分泌物和排泄物dna样品的基因组分析,这是由于细菌和病毒dna中不存在alu、mir和sva逆转录转座子,使细菌和病毒dna对人体dna分析的干扰減至最低。
本发明所述的多功能almivascan方法平台,是通过ite-pcr捕获alu、mir和sva逆转录转座子邻近区域的人类基因组dna序列,再以mps测序并进行基因组序列分析。平台具三重优点:第一,本发明所发现的alu、mir和sva插入位点,均富集于基因组变异附近,如单核苷酸多态性(snps)、微卫星(msts)、种系拷贝数变异(cnvgs)和体细胞拷贝数变异(cnvts)等邻近序列中;这些基因组变异可用于区分不同人类受试者的遗传性状,以及检测带有遗传变异并显示为疾病基因型(如癌症或癌前基因型)的细胞。第二,经almivascan平台产出的ite-pcr扩增子,于凝胶电泳及溴化乙锭染色后,呈光亮弥散状模式,这表示扩增产物含高量ite基因组片段,可用于后续序列分析。第三,该平台提供了一种人类基因组序列的分析工具,适用于分析从血浆、痰液、粪便,及任何受细菌和病毒污染的人类分泌物和排泄物中分离出来的人类dna样品。
提供了一种方法,用于鉴定人类基因组dna中一或多种的基因组变异。该方法包含制备测定用混合物,内含:人类基因组dna的测试样品;两条或以上引物;四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶;和缓冲液。所述的两条或以上引物中的每一个均包含基于人类基因组转座子(te)或其他重复元件序列的共有序列,而所述的人类基因组转座子通常在微生物dna中不会找到的。然后,所述方法包含把上述测定混合物进行聚合酶链式反应(pcr),生成一含人类基因组转座子间(ite)区段的扩增子阵列。
提供一种方法,用于鉴定与性状或疾病关联的一或多种基因组变异体。该方法包含制备第一和第二测定混合物,第一测定混合物内含:第一测试样品;两条或以上引物;四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶;和缓冲液。第一测试样品包含人类基因组dna,是取自具有某特定性状或疾病的受试者。所述的两条或以上引物中的每一条均包含基于人类基因组转座子(te)或其它重复元件序列的共有序列,而所述的人类基因组转座子te或其他重复元件通常是微生物dna中不会找到的。所述第二测定混合物内含:第二测试样品;两条或以上引物;四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶;和缓冲液。第二测试样品同样包含人类基因组dna,但取自没有所述特定性状或疾病的另一受试者,所用的两条或以上引物,其序列与第一测定混合物中所使用的一样。然后,把所述第一和第二测定混合物分别进行聚合酶链式反应(pcr),并分别生成以第一测试样品为模板的基因组转座子间(ite)序列扩增子,与及以第二测试样品为模板的基因组转座子间(ite)序列扩增子。
提供了一种试剂盒,用于对内含人类基因组dna的测试样品进行聚合酶链式反应(pcr)。该试剂盒包含第一寡核苷酸引物和第二寡核苷酸引物,其中,所述寡核苷酸引物包含选自以下的一核苷酸序列:agcttgcagtgagctgagat(seqidno:6),gtccgcagtccggcctgggc(seqidno:7),gatagcgccactgcagtcc(seqidno:8),agccgagatggcagcagta(seqidno:11),及accagagacctttgttcact(seqidno:12),而第一和第二寡核苷酸引物的序列彼此不同。该试剂盒还附有说明书,指示如何使用第一和第二寡核苷酸引物和人类dna测试样品进行pcr反应,用于检测样品中的人类基因组变异。
其它目的和特点,部分为显而易见,部分将在下文阐述。
附图说明
以下将详细说明本发明的附图和实施例:
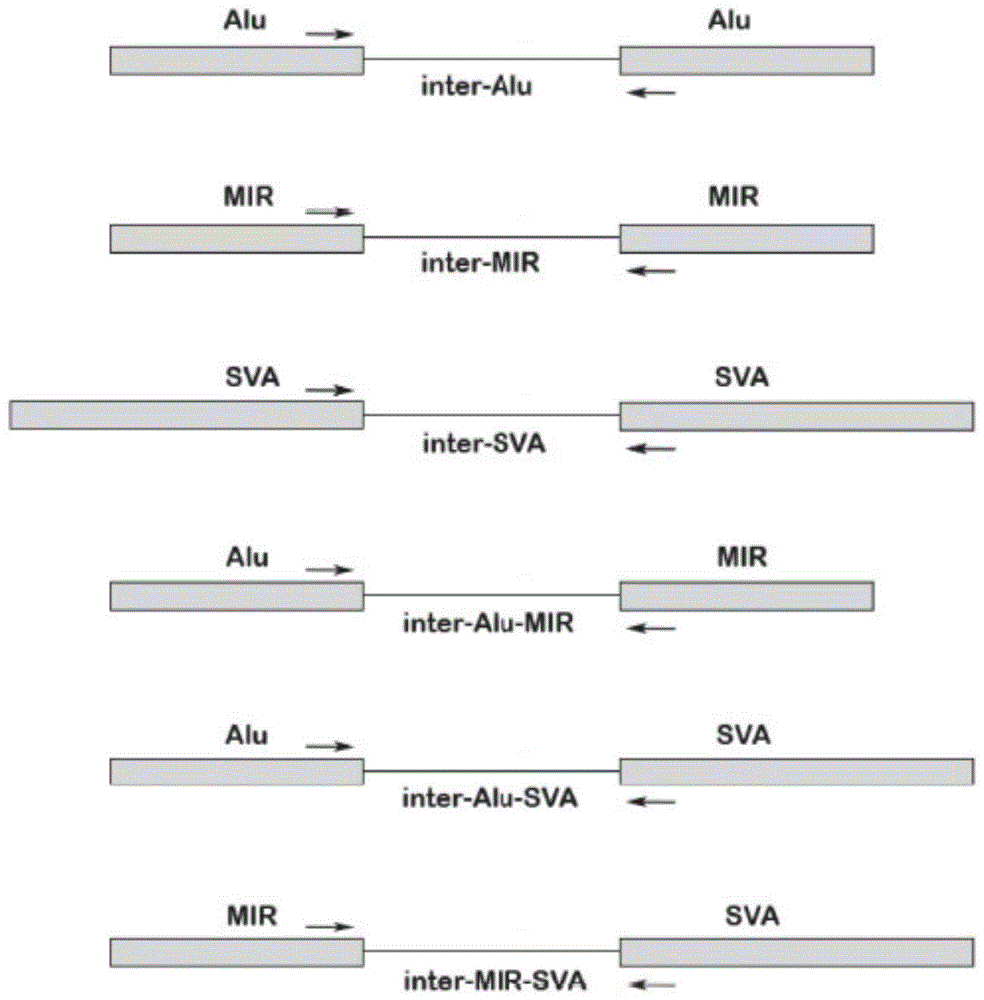
图1为转座子间pcr扩增说明。图中显示扩增点为两个相邻转座子(tes)(以两个长方形代表)之间的转座子间序列区段(以行线代表),使用两条te-共有序列基引物,其中一条用于左边的te,另一条则是右边的te(扩增方向以箭头表示)。从上至下是:两个alu元件之间的alu间区段的pcr;两个mir元件之间的mir间区段的pcr;两个sva元件之间的sva间区段的pcr;alu-和mir-元件之间的alu-mir间区段的pcr;alu-和sva-元件之间的alu-sva间区段的pcr;以及mir-和sva-元件之间的mir-sva间区段的pcr。
图2显示扩增点为两个相邻转座子(tes)(以两个长方形代表)之间的转座子间(ite)序列区段(以行线代表),使用两条te-共有序列基引物,一条用于左边te,另一条用于右边te(扩增方向以箭头表示)。在pcr扩增过程中,所用的每条引物可以是通过te头部进行链延伸的h-型,或是通过te尾部进行链延伸的t-型。从上至下是:“头对头”扩增;“尾对尾”扩增;“头对尾”扩增;及“尾对头”扩增。这四种扩增模式均可以应用于扩增图1所示的六种转座子间序列。
图3显示不同逆转录转座子群组在离开千人基因组计划(snp1ks)中各单核苷酸多态性(snps)位点不同距离处的密度变异。是按各逆转录转座子群组在离开“snp1k”数据库中各snp不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组在基因组的平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算1,000,000个snps(n=100x)的随机模拟分布(northetal2002)计算。如一些逆转录转座子亚家族有着非常近似的snps密度分布的,被分组在一起:在此基础上,六个sva亚家族会被分成sva_ef组(sva_e和sva_f)、sva_cd组(sva_c和sva_d)和sva_ab组(sva_a和sva_b);而aluy亚家族则分为aluy_young组(aluya5、aluyb8、aluyb9、aluyh和aluyk12),aluy_old组(aluy、aluyc、aluyc3和aluyk4)及alu_old组(alujb、alujr4和flam_a)。
图4显示不同逆转录转座子群组在离开人类微卫星(msts)位点不同距离处的密度变异。是按各逆转录转座子群组在离开各mst不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组的基因组平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算基因组中41,572个msts(n=100x)的随机模拟分布(northetal2002)计算。各逆转录转座子亚家族是按图3所述分组。
图5显示不同逆转录转座子群组在离开人类短种系拷贝数变异(shortcnvgs,5bp<cnvgs≤158bp)位点不同距离处的密度变异。是按各逆转录转座子群组在离开各shortcnvg不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组的基因组平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算基因组中157,793个shortcnvgs(n=100x)的随机模拟分布(northetal2002)计算。各逆转录转座子亚家族是按图3所述分组。
图6显示不同逆转录转座子群组在离开人类中长种系拷贝数变异(midcnvgs,158bp<cnvgs≤15,848bp)位点不同距离处的密度变异。是按各逆转录转座子群组在离开各midcnvg不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组的基因组平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算基因组中350,037个midcnvgs(n=100x)的随机模拟分布(northetal2002)计算。各逆转录转座子亚家族是按图3所述分组。
图7显示不同逆转录转座子群组在离开人类长种系拷贝数变异(longcnvgs,cnvgs>15848bp)位点不同距离处的密度变异。是按各逆转录转座子群组在离开各longcnvg不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组的基因组平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算基因组中256,085个longcnvgs(n=100x)的随机模拟分布(northetal2002)计算。各逆转录转座子亚家族是按图3所述分组。
图8显示不同逆转录转座子群组在离开人类体细胞拷贝数变异(cnvts)位点不同距离处的密度变异。是按各逆转录转座子群组在离开各cnvt不同距离处(单位为kb,以100bp递增)的平均密度绘制。箭头所指位置显示各逆转录转座子群组的基因组平均密度,虚线以上的密度值显示高于平均密度值,显著性差异达p<0.01。所述的平均密度值是通过蒙特卡罗(montecarlo)程序运算基因组中985,038个cnvts(n=100x)的随机模拟分布(northetal2002)计算。各逆转录转座子亚家族是按图3所述分组。
图表9列出在离开各种形式的人类基因组多态性不同距离处,不同逆转录转座子组的富集(达p<0.01)情况。各逆转录转座子亚家族是按图3所述分组。
图10为ite-pcr扩增子的凝胶电泳图,是利用不同组合的ite-pcr引物和12.5ng人类白血细胞dna为模板进行扩增。泳道a是dna分子量(m.w.)标记;泳道b-p是使用各种ite-pcr引物组合获得的ite-pcr扩增子电泳图,下列表一列出各引物组合,其序列则见于实施例7中:
表一
图11为ite-pcr扩增子的凝胶电泳图,是利用不同组合的ite-pcr引物和12.5ng人类痰液dna为模板进行扩增。泳道a是dna分子量(m.w.)标记;泳道b-p是使用了表一所列的各引物组合而生成的ite-pcr扩增子电泳图。
图12为ite-pcr扩增子的凝胶电泳图,是利用不同组合的ite-pcr引物和125ng人类粪便dna为模板进行扩增。粪便取样需经过72小时无肉饮食后。泳道a是dna分子量(m.w.)标记;泳道b-p是使用了表一所列的各引物组合而生成的ite-pcr扩增子电泳图。
发明详述
在详述本发明前,应先理解本发明在不同情况下可以变动,因此并不限于使用所述的特定ite-pcr引物来扩增ite序列片段;或ite-pcr反应中所述的试剂或程序。还应该理解的是,此处所使用的术语仅为了说明本发明的具体实施例,并不意味着仅限于所述内容。另外,在详述本发明的实施例前,阐述相关定义将有助对本发明的理解。所阐述的术语定义仅适用于本专利,在其它地方使用时,相同术语的定义或有所不同,如在科学文献或其它专利或应用上。另外,示例时,它们仅用作说明而不具限制性。
必须注意的是,除非另有说明,说明书及以下权利要求中所使用的单数字眼“一”、“一个”和“该”亦含复数之意。例如引用“引物”时,可能意指两条或以上引物组成。
术语“包含”、“包括”和“具有”意指包括在内,并可能包括已被列出以外的要素。
无论在说明及权利要求中,本发明所用术语,将依据以下所订定义。
术语“pcr产物”意指经转座子间pcr生成及使用乙醇或其它纯化试剂盒处理并去除任何过剩引物、酶、矿物油、甘油和盐的pcr产物。
术语“转座子间序列”,即“ite序列”,可以是位于两个alu元件之间、两个mir元件之间、两个sva元件之间、一个alu元件和一个mir元件之间、一个alu元件和一个sva元件之间,或一个mir元件和一个sva元件之间的dna序列片段(图1)。ite-pcr意指采用基于alu-、mir-和/或sva-共有序列设计的引物对任何ite序列片段进行pcr扩增。由于alu和mir元件在人类基因组中广泛存在,而alu元件和mir元件分别富集于基因区和近端区序列中。同时使用alu-和mir-共有序列基的引物进行itepcr,将可覆盖人类基因组的重要不同部分,尤其是基因组中具高功能性的基因区和近端区序列。
术语“质量”是指ite-pcr扩增子的两个基本属性,就是:仅需使用纳克量人类dna样品作pcr扩增,以及扩增子的多样性。所产生的扩增子量必须足以用来进行mps测序,这可通过紫外线(uv)照射经溴化乙锭染色电泳后dna扩增子的亮度得知;而扩增子的多样性也必须足以使mps分析达至高效及高产能,这可通过凝胶电泳图中扩增子呈弥散状而非明显条带状来显示扩增序列的多样性。
术语“纳克量基因组dna”是指ite-pcr所需的亚微克量dna,用作生成ite-pcr扩增子及后续的大规模并行测序。
术语“热稳定dna聚合酶”是指ite-pcr中使用的dna聚合酶,可以是taq聚合酶、kod聚合酶或其它用于dna扩增的聚合酶。
术语“扩增方向”意指pcr扩增中的链延伸方向,是将pcr引物通过转座子(te)的5'端(头部)或3'端(尾部)序列延伸。“头型”(h-型)引物会经过te的5'头部方向进行链延伸,而“尾型”(t-型)引物会经te的3'尾部方向延伸。
术语“头对头”意指被扩增的ite序列是位于一te5'端及其相邻te5'端之间的ite片段(见图2,顶线)。
术语“尾对尾”意指被扩增的ite序列是位于一te3'端及其相邻te3'端之间的ite片段(见图2,第二线)。
术语“头对尾”意指被扩增的ite序列是位于一te5'端及其相邻te3'端之间的ite片段(见图2,第三线)。
术语“尾对头”意指被扩增的ite序列是位于一te3'端及其相邻te5'端之间的ite片段(见图2,第四线)。
术语“大规模并行测序”(“mps”)涵盖了几种高通量dna测序技术,亦被称为“新一代测序”(“ngs”)。这些技术采用小型化和并行化平台进行1-100百万次短读长测序(50-400碱基),许多mps平台有着不同的工程设置和测序化学。然而,它们均运用了大规模并行测序的技术范式通过空间分离,将由克隆扩增dna模板或流通池中的单dna分子;设计上与sanger测序(也被称为毛细管测序或第一代测序)有着明显差异,sanger测序是基于将个别测序反应中产生的链终止物进行电泳分离并读取其碱基序列。
术语“扩增子”意指从天然或人工扩增生成的一段dna。最常见是通过聚合酶链式反应(“pcr”)。通常,选择性扩增某特定一组ite片段,并对其凝胶电泳图中呈条带状进行片段大小评估。通过多引物ite-pcr扩增,可产生大量不同大小的ite-扩增子,它们是从集有alu-、mir-和/或sva-元件的基因组dna区域扩增,并且不同元件彼此之间的距离属pcr可扩增范围内。在本发明中,利用多引物ite-pcr扩增基因组不同区域中的相邻alu、mir和/或sva元件之间区域,以头对头、尾对尾、头对尾和尾对头方向延伸,所获取的扩增产物经电泳分离及溴化乙锭染色。若显示为条带状的话,这显示获扩增的转座子间区段数量相对较少;而呈弥散模式(非条带状)则显示获扩增的转座子间区段数量非常庞大。
为测试和验证almivascan的捕获潜力,特别是针对富含基因组多态性的ite序列,本发明已对alu、mir和sva元件位置及其相关基因组多态性位点进行了分析,如snps、msts、cnvgs和cnvts等基因组多态性位点。如图3-9所示,于各snp、mst、cnvg及cnvt多态性位点约3kb范围内pcr可扩增距离的序列当中,明显富含alu、mir和sva元件,显著性达p<0.01,这表示alu、mir和sva的插入可能是诱发snp、mst、cnvg和cnvt多态性的重要因素。在这方面,已知snps与alu元件(ngandxue2006)及一些人类性状和疾病(batzeranddeininger2002;claussnitzeretal2015;fujimoto2009;kim2012etal;kumar,yang,xueetal2015;lo,lau,xueetal2004;mbareketal2016;medlandetal2016;mei,ding,xueetal2011;sturmetal,2005;walkeretal2003;zhao,xu,xueetal2009)相关;而msts占基因组可变基因座数目的2.7%,远高于snps,能调节基因表达,并且与人类性状相关(payseuretal2011;sawayaetal2012;bagshawetal2017);另外,cnvgs与人类身高和癌症易感相关(lietal2010;ding,tsang,xueetal2014);及cnvts在枢神经系统发育、衰老和癌症中扮演着重要角色(iourovetal2008;schlienandmalkin2009)。因此,应用alu-、mir-和sva-共有序列基引物进行的pcr扩增,将提供一种有效工具,用于捕获富集基因组多态性位点的ite序列以进行测序,图3-9显示了丰富的基因组多态性,包含可能与各种人类基础性状相关多态性。
为了测试almivascan平台用于扩增庞大数量及多样性扩增子的潜力,而所述扩增是基于alu-、mir-和sva-逆转录转座子共有序列设计的ite-pcr引物。则必须寻找一组具说明作用的alu-、mir-和sva-序列基引物,不但可以生成大量扩增子,使溴化乙锭染色显得明亮;并且包含极高的序列多样性扩增子,令到凝胶电泳图中呈现弥散而非条带状模式。就此而言,图10-12中所示的凝胶电泳图验证了采用不同的alu-、mir-和sva-序列基pcr引物组合所产生的ite-pcr扩增子,其凝胶电泳图均显示具有明亮的溴化乙锭染色及呈广泛弥散状模式的泳道,这表示出当中含有大量可供mps序列分析的多样性基因组ite片段。
所述方法用于鉴定来自不同或同一受试者的不同dna样本之间的遗传差异,这些遗传差异是存于相关基因组ite序列片段中。步骤包括:(a)选择一组配对样品,其中某特定基因型只能在第一个样品中表达,而不能在第二个样品中表达;(b)选定以alu-、mir-和sva-共有序列设计的ite-pcr引物,并对该组配对样品中的每个样品进行ite-pcr,扩增一阵列基因组ite序列区段;(c)采用mps技术对每组配对样品中的基因组ite序列扩增片段进行测序,描绘成为“almivascan”;(d)把每组配对样品的mps结果(亦即almivascans)存储为计算机可读格式;(e)从mps结果中鉴定相关的基因组ite序列片段,亦即almivascans,用于识别该组配对样品之间某基因序列或结构的差异,当中包括但不限于单核苷酸多态性(snp)、单核苷酸变异(snv)、微卫星(mst)或拷贝数变异(cnv)。
由于alu元件仅在灵长类动物基因组中被发现,mir元件仅在哺乳动物基因组中被发现,及sva元件仅在人科动物基因组中被发现,因此“almivascan”平台在人类基因组dna样品的应用将不会受样品中的微生物dna或植物dna污染所干扰。另外,若受试者在受试dna取样前72小时开始不进食任何含牛肉、猪肉、羊肉或其它含哺乳动物或脊椎动物成分的食品,那取自该人类样品中的基因组dna受哺乳动物dna干扰的机会会很小。综合所述的,“almivascan”分析不仅适用于人类受试者取自血液或组织的dna样品,也适用于取自粪便、痰液或口腔等分泌物和排泄物的dna样品。鉴于此,本发明一个实施例中的dna样品是来自人类白血球细胞,另一个实施例中的dna样品是来自人类痰液,又一个实施例中的dna样品是来自人类粪便。而痰液dna样本经常受存于肺部和呼吸道的微生物污染,粪便dna样本则常受胃肠系统中的微生物污染。
almivascan测序平台的核心优势源于人类基因组中具有大量alu和mir插入位点,它们分别富集于基因区序列和近端区序列中。要是采用alu-和mir-共有序列基的引物进行ite-pcr,将可以从人类基因组中扩增庞大数量的ite-扩增子。虽然sva插入位点数目相对较少,但除了sva间序列之外,也可通过扩增alu-sva间和mir-sva间序列,加强sva相邻序列的扩增(图1)。此优点亦可以应用于扩增其它插入位点较少而不出现于在细菌和病毒中的一般转座子(或gte),例如l1等,只要微生物dna中不存在任何gte序列,便可以通过使用alu-、mir-和gte-共有序列基的ite-pcr引物组合扩增ite序列。其中除了gte间序列之外,gte-相邻序列的扩增还可以通过捕获alu-gte间及mir-gte间序列进行。
提供一种序列分析的方法。所述方法使用多种转座元件(ite)-pcr引物来扩增ite序列,用于后续测序,及发现和/或分析人类基因组序列和结构变异与个体或细胞性状之间的关联,包括患病状态、疾病易感性和药物反应。步骤包括:
(a)用以下组分的pcr反应混合物进行ite-pcr:
i.两条或以上alu-、mir-和/或sva-共有序列基ite-pcr引物;
ii.人类基因组dna样品;及
iii.pcr扩增混合物,包括:四种分别含a、g、t和c碱基的游离脱氧核苷酸三磷酸、
热稳定dna聚合酶和缓冲液;
(b)在pcr仪中,对pcr反应混合物进行ite-pcr扩增程序,经一段时间后完成,产生一阵列alu间、mir间、sva间、alu-mir间、alu间-sva和/或mir-sva间序列的扩增子;
(c)对alu间、mir间、sva间、alu-mir间、alu-sva间及mir-sva间序列阵列进行序列分析,比较来自相同或不同受试者的测试dna样品和对照dna样品的序列分析结果,以便确定基因组序列变异与个体或细胞性状之间的相关性。
设计一条或以上ite-pcr引物,除了基于alu、mir和sva逆转录转座子以外,还可以基于其它不出现于细菌和病毒dna的逆转录转座子或重复序列的共有序列。
dna样品可以从人类白血球细胞中制备。
dna样品可以从人类痰液中制备。
dna样品可以从人类粪便中制备。
dna样品可以从人类血浆中制备。
dna样品可以从人类血清中制备。
dna样品可以从人类尿液中制备。
dna样品可以从人类唾液制备。
除了白血球细胞、血浆和血清之外,dna样品可以从任何人类组织中制备。
除痰液、粪便、尿液和唾液以外,dna样品可以从任何分泌物或排泄物中制备。
在任何方法中,相关的ite间序列多态性可以与个体性状或细胞性状或疾病找到关联,如下所示:
(a)选择一组配对样品,其中一个样品能够表达某性状,而另一个则不能表达;
(b)为所述配对样品中的每一个样品产生一ite序列区段阵列;
(c)使用大规模并行测序技术对从该配对样品所产生的ite序列区段阵列中每一个序列进行测序,生成一大规模并行ite序列阵式;
(d)将每组配对样品的大规模并行ite序列阵式转换成计算机可读格式;和
(e)于检测到配对样品之间的可识别的遗传变异的大规模并行ite序列阵式中,识别出相关ite序列元件,而将所述相关ite序列元件鉴定为与该遗传性状有关联的序列。
所述方法可进一步包括,于大规模并行ite序列阵式中识别出相关的ite序列,其中配对样本间的可识别的遗传变异由该变异碱基的读取质量得分阈值(thresholdreadqualityscore)表示。
所述方法可进一步包括,于大规模并行ite序列阵式中识别出相关的ite序列,其中配对样本间的可识别的遗传变异是由比对该遗传变异与表观性状的体内平衡标记物的关键性阈值表示的。
所述方法可进一步包括,于大规模并行ite序列阵式中识别出相关的ite序列,其中配对样本间的可识别的遗传变异是由比对该遗传变异与性状的严重程度的阈值表示的。
所述方法可进一步包含以下步骤:选择杂合性丢失为该配对样品之间的可识别遗传变异。
所述方法可进一步包含以下步骤:选择体细胞的插入/缺失为该配对样品之间的可识别遗传变异。
所述方法可进一步包含以下步骤:选择单核苷酸变异(“snv”)为该配对样品之间的可识别遗传变异。
所述方法可进一步包含以下步骤:选择移码变异为该配对样品之间的可识别遗传变异。
可使用新型dna引物,序列为:[5'-agcttgcagtgagctgagat-3'](seqidno:6)在本发明任何方法中作为ite-pcr引物之一。
可使用新型dna引物,序列为:[5’gtccgcagtccggcctgggc-3’](seqidno:7)在本发明任何方法中作为ite-pcr引物之一。
可使用新型dna引物,序列为:[5’gatagcgccactgcagtcc-3’](seqidno:8)在本发明任何方法中作为ite-pcr引物之一。
可使用新型dna引物,序列为:[5’agccgagatggcagcagta-3’](seqidno:11)在本发明任何方法中作为ite-pcr引物之一。
可使用新型dna引物,序列为:[5’accagagacctttgttcact-3’](seqidno:12)在本发明任何方法中作为ite-pcr引物之一。
提供了一种方法,用于鉴定人类基因组dna中的一或多种基因变异。该方法包含制备测定混合物,所述测定混合物当中包括:含人类基因组dna的测试样品;两条或以上引物;四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶和缓冲液。所述两条或以上引物中每一条引物均包含基于人类基因组转座子(te)或其它重复元件的共有序列,而该等te或其它重复元件通常不会在细菌和病毒dna中出现。所述方法还包括对测定混合物进行聚合酶链式反应(pcr),产生一含基因组转座子间(ite)区段的扩增子阵列。
所述方法可进一步包括应用高通量dna测序技术对所产生的扩增子进行测序。
所述方法可进一步包括将扩增子序列与对照dna样品的相同人类基因组区域序列进行比较,以鉴定测试样品和对照dna样品之间的一或多种基因组变异。
所述对照dna样品可以是取自人类受试者的基因组dna样品,并通过所属领域的任何已知方法测序。又或是对照dna样品可以是含已知的人类基因组dna序列。
提供了一种方法,用于鉴定与性状或疾病相关的一或多种基因组变异。该方法包含制备第一和第二测定混合物。第一测定混合物中含有:包含人类基因组dna的第一测试样品,其中,第一测试样品是从带有某特定性状或疾病的人类受试者中获取的;另外,第一测定混合物还包括两条或以上引物,其中每一条引物包含基于人类基因组转座子(te)或其它重复元件的共有序列,而所述te或其它重复元件通常不会在微生物dna中找到;第一测定混合物还包含四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶和缓冲液。第二测定混合物中含有:含有人类基因组dna的第二测试样品,其中,第二测试样品是从没带有某特定性状或疾病的人类受试者组织中获取;第二测定混合物还含有所用的两条或以上引物,其序列与第一测定混合物中的两条或以上引物相同;另外,第二测定混合物亦包括四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps);热稳定dna聚合酶和缓冲液。所述方法包含分别对第一和第二测定混合物进行聚合酶链式反应(pcr),分别产生第一测试样品基因组转座子间(ite)区段的扩增子阵列和第二测试样品基因组转座子间(ite)区段的扩增子阵列。
所述方法可进一步包括应用高通量dna测序技术分别对第一和第二扩增子阵列进行测序。
所述方法可进一步包括将第一扩增子阵列的序列与第二扩增子阵列的序列进行比较,以鉴定第一和第二测试样品之间的一或多种基因组变异。要是该基因组变异仅发现于第一测试样品中,但不存于第二测试样品中,这表明它可能与性状或疾病相关联。
提供了一种试剂盒,用于对含有人类基因组dna的测试样品进行聚合酶链式反应(pcr)。该试剂盒包含第一寡核苷酸引物和第二寡核苷酸引物,其中,寡核苷酸引物包含选自以下一条的核苷酸序列:agcttgcagtgagctgagat(seqidno:6),gtccgcagtccggcctgggc(seqidno:7),gatagcgccactgcagtcc(seqidno:8),agccgagatggcagcagta(seqidno:11)及accagagacctttgttcact(seqidno:12),而所述第一和第二寡核苷酸引物的序列彼此不同。该试剂盒还附有说明书,指示如何使用第一和第二寡核苷酸引物和进行pcr反应,用于鉴定测试样品中人类基因组dna的基因组变异。
所述试剂盒还可选择包含其它pcr反应所需的组分,如热稳定dna聚合酶;四种分别含腺嘌呤(a)、胞嘧啶(c)、鸟嘌呤(g)和胸腺嘧啶(t)碱基的游离脱氧核苷酸三磷酸(dntps)和/或缓冲液。
在任何所述方法或试剂盒中,基因组变异将包括单核苷酸多态性(snp)、微卫星(mst)、种系拷贝数变异(cnvg)(如短cnvg、中长cnvg、长cnvg或其任何组合)、体细胞拷贝数变异(cnvt)或其任何组合。
测试样品可以包含任何人体组织、分泌物或排泄物。
测试样品、第一测试样品、和/或第二测试样品可包含人类白血球细胞。
测试样品、第一测试样品、和/或第二测试样品可包含人类痰液。
测试样品、第一测试样品、和/或第二测试样品可包含人类粪便。而收集该测试样品、第一测试样品、和/或第二测试样品中的人类粪便前,受试者需经过72小时无进食任何源自哺乳动物或脊椎动物的肉或组织。
测试样品、第一测试样品、和/或第二测试样品可包含人类血浆。
测试样品、第一测试样品、和/或第二测试样品可包含人类血清。
测试样品、第一测试样品、和/或第二测试样品可包含人类尿液。
测试样品、第一测试样品、和/或第二测试样品可包含人类唾液。
测试样品、第一测试样品、和/或第二测试样品可包含人类白细胞、人类痰液、人类粪便、人类血浆、人类血清、人类尿液和人类唾液的任何组合。
测试样品、第一测试样品、和/或第二测试样品可包含亚微克量的人类dna。
测试样品、第一测试样品和/或第二测试样品可进一步包含微生物dna、植物dna、非脊椎动物dna或其任何组合。
测试样品、第一测试样品和/或第二测试样品可进一步包含微生物dna、植物dna,或微生物dna和植物dna。人类粪便样品通常含有大量的微生物和植物dna。
测试样品、第一测试样品和/或第二测试样品可进一步包含微生物dna。
存在于测试样品、第一测试样品和/或第二测试样品中的微生物dna和/或植物dna,甚至在高量的情况下,都不会干扰样品的测定结果。就是说,测试样品、第一测试样品或第二测试样品中的微生物和/或植物dna与人类dna的比例可介乎0:1和100:1之间,如1:1或50:1。
所述两条或以上引物中的每一条引物可包含人类基因组转座子(te)共有序列,其中的te通常不出现于微生物dna中。
例如,所述两条或以上引物中的每一条引物可包含alu元件基共有序列、mir元件基共有序列或sva元件基共有序列。
例如,测定混合物、第一测定混合物和/或第二测定混合物中可包含一条基于alu元件共有序列设计的引物及一条基于mir元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少一条基于alu元件共有序列设计的引物及至少一条基于sva元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少一条基于mir元件共有序列设计的引物和至少一条基于sva元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少两条基于mir元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少两条基于sva元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少一条基于alu元件共有序列设计的引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含两条或以上基于alu元件共有序列设计的引物。
优选地,测定混合物、第一测定混合物和/或第二测定混合物中含有两条或以上基于alu元件共有序列设计的引物,及一条或以上基于mir元件共有序列设计的引物。
更优选地,测定混合物、第一测定混合物和/或第二测定混合物中含有两条或以上基于alu元件共有序列设计的引物,及一条或以上基于sva元件共有序列设计的引物。
最优选地,测定混合物、第一测定混合物和/或第二测定混合物中含有至少四条或以上基于alu元件共有序列设计的引物,至少一条或以上基于mir元件共有序列设计的引物,及至少一条或以上基于sva元件共有序列设计的引物。
至少一条引物可包含alu元件基共有序列。
至少一条引物包含基于alu元件的共有序列,该引物包含选选自以下一条的核苷酸序列:tggtctcgatctcctgacctc(seqidno:2),gagcgagactccgtctca(seqidno:3),tgagccaccgcg(seqidno:4),agcgagactccg(seqidno:5),agcttgcagtgagctgagat(seqidno:6),gtccgcagtccggcctgggc(seqidno:7),andgatagcgccactgcagtcc(seqidno:8)。
至少一条引物包含基于alu元件共有序列,该引物包含核酸序列:agcttgcagtgagctgagat(seqidno:6)
至少一条引物包含基于alu元件的共有序列,该引物包含核苷酸序列:gtccgcagtccggcctgggc(seqidno:7)。
至少一条引物包含基于alu元件的共有序列,该引物包含核苷酸序列是:gatagcgccactgcagtcc(seqidno:8)。
至少一条引物可包含基于mir元件的共有序列。
至少一条引物包含基于mir元件的共有序列,该引物包含核苷酸序列:gtgacttgctcaaggt(seqidno::9)和gcctcagtttcctcatc(seqidno:10)。
至少一条引物可包含基于sva元件的共有序列。
至少一条引物包含基于sva元件的共有序列,该引物包含核苷酸序列:agccgagatggcagcagta(seqidno:11)。
至少一条引物包含基于sva元件的共有序列,该引物包含核苷酸序列:accagagacctttgttcact(seqidno:12)。
theassaymixture,thefirstassaymixture,and/orthesecondassaymixturecancomprisethreeormoreprimers.
测定混合物、第一测定混合物和/或第二测定混合物中可包含三条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含四条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含五条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含六条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含七条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含八条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含九条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含十条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含十一条或以上引物。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少五条引物,其中所述引物包含seqidno:2、3、4、5和9的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少六条引物,其中所述引物包含seqidno:2、3、4、5、11和12的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少五条引物,其中所述引物包含seqidno:2、3、4、5和8的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少五条引物,其中所述引物包含seqidno:2、3、4、5和7的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少五条引物,其中所述引物包含seqidno:2、3、4、5和6的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少六条引物,其中所述引物包含seqidno:2、3、4、5、9和10的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少十条引物,其中所述引物包含seqidno:2、3、4、5、6、7、8、9、11和12的核苷酸序列。
测定混合物、第一测定混合物和/或第二测定混合物中可包含至少十一条引物,其中所述引物包含seqidno:2、3、4、5、6、7、8、9、10、11和12的核苷酸序列。
所述引物组优选包含至少一条头向型(h-型)引物,该h-型引物将向转座子或其它重复元件的头方以外延伸;及至少一条尾向型(t-型)引物,该t-型引物则向转座子或其它重复元件的尾方以外延伸。
基因组转座子间(ite)区段可包括alu间区段、mir间区段、sva间区段、alu-mir间区段、alu-sva间区段和mir-sva间区段,或其任何组合。
高通量dna测序可包括大规模并行测序。
大规模并行测序可产生大规模并行的转座子间(ite)序列阵式。
所述方法可进一步包括将测序获得的数据转换成计算机可读格式,用于比较扩增子与对照样品dna的序列。
所述方法可进一步包括将测序获得的数据转换成计算机可读格式,用于比较第一和第二阵列扩增子的序列。
所述方法可进一步包括从大规模并行的转座子间(ite)区段序列阵式中,识别出相关的ite基因区段,其中第一和第二测试样品之间存在的遗传变异由一个或多个变异碱基的读取质量得分阈值表示。
所述方法可进一步包括从大规模并行阵式的转座子间(ite)区段序列阵式中,识别出相关的ite基因区段,其中第一和第二测试样品之间存在的遗传变异,是通过对某特定性状或疾病标记物的比对,以重要性阈值表示。
所述方法可进一步包括从大规模并行阵式的转座子间(ite)区段序列阵式中,识别出相关的ite基因区段,其中第一和第二测试样品之间存在的遗传变异,是通过对某特定性状或疾病的严重度比对,以阈值表示。
第一和第二测试样品之间的遗传变异包括杂合性丢失。
第一和第二测试样品之间的遗传变异包括体细胞的插入/缺失。
第一和第二测试样品之间的遗传变异包含单核苷酸多态性。
提供了一寡核苷酸。该寡核苷酸包含选自下列一条的核苷酸序列:agcttgcagtgagctgagat(seqidno:6),gtccgcagtccggcctgggc(seqidno:7),gatagcgccactgcagtcc(seqidno:8),agccgagatggcagcagta(seqidno:11),及accagagacctttgttcact(seqidno:12)。
提供了对包含人类基因组dna的测试样品进行聚合酶链式反应(pcr)的一试剂盒,所述试剂盒包含:
第一寡核苷酸引物和第二寡核苷酸引物,其中所述寡核苷酸引物包含选自以下的一核苷酸序列:agcttgcagtgagctgagat(seqidno:6),gtccgcagtccggcctgggc(seqidno:7),gatagcgccactgcagtcc(seqidno:8),agccgagatggcagcagta(seqidno:11),及accagagacctttgttcact(seqidno:12);以及第一和第二寡核苷酸引物彼此不同;和
使用第一和第二寡核苷酸引物在pcr反应中检测包含人基因组dna的测试样品中人基因组dna中基因组变异的说明书。
本发明已经详细描述,很明确地,在不脱离本发明范围所附权利要求限定的情况下,可以进行修改和变更。
实施例
以下的非限制性实施例将进一步具体地说明本发明。
实施例一
单核苷酸多态性位点(snps)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与snp1k数据库中的snps位置进行比较,从而检测所述逆转录转座子和snps之间的潜在相关性。图表3和图9总结显示了个别元件于离开snp1k中各snp位置不同相距范围内的平均密度,包括sva_cd和sva_ab于相距5kb范围内,mir于相距1-5kb范围内,aluy-young于相距0.25kb范围内,aluy-old于相距3.7kb范围内和alus于相距0.95kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的100百万个snps比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例二
微卫星(msts)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与mst数据库中的msts位置进行比较,从而检测所述逆转录转座子和msts之间的潜在相关性。图表4和图9总结显示了个别元件于离开各mst位置不同相距范围内的平均密度,包括mir于相距1.65-5kb范围内,aluy_old于相距0.3–0.75kb和1.05–2.35kb范围内,alus于相距0.2–5kb范围内,及alu_old于相距5kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的所有msts比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例三
短种系拷贝数变异(shortcnvgs)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与devars数据库中的shortcnvgs(5bp<cnvg≤158bp)所在位置进行比较,从而检测所述逆转录转座子和shortcnvgs之间的潜在相关性。图表5和图9总结显示了个别元件于离开各shortcnvg位置不同相距范围内的平均密度,包括sva_ef、sva_cd、aluy_young、aluy_old、alus和alu_old于相距5kb范围内,sva_ab于相距0.6–2.95kb范围内,及mir于相距1.4–5kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的所有shortcnvgs比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例四
中长种系拷贝数变异(midcnvgs)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与devars数据库中的midcnvgs(158bp<cnvg≤15848bp)所在位置进行比较,从而检测所述逆转录转座子和midcnvgs之间的潜在相关性。图表6和图9总结显示了个别元件于离开各midcnvg位置不同相距范围内的平均密度,包括sva_ef和aluy_young于相距5kb范围内,sva_cd于相距0.7–5kb范围内,及aluy_old于相距0.15–5kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的所有midcnvgs比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例五
长种系拷贝数变异(longcnvgs)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与devars数据库中的longcnvgs(cnvg>15848bp)所在位置进行比较,从而检测所述逆转录转座子和longcnvgs之间的潜在相关性。图表7和图9总结显示了个别元件于离开各longcnvg位置不同相距范围内的平均密度,包括sva_ef于相距3.75–5kb范围内,sva_cd于相距4.25-5kb范围内,aluy_young于相距0.1–5kb范围内,aluy_old于相距0.9–5kb范围内及alus于相距1.7–5kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的所有longcnvgs比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例六
体细胞拷贝数变异(cnvts)与alu、mir和sva逆转录转座子的相对位置。通过ucsc人类参考基因组hg19(smitetal2010)浏览器的repeatmasker来查找alu、mir和sva逆转录转座子的位置,并与cosmic数据库(forbesetal2015)中的cnvts所在位置进行比较,从而检测所述逆转录转座子和cnvts之间的潜在相关性。图表8和图9总结显示了个别元件于离开各cnvt位置不同相距范围内的平均密度,包括sva_cd于相距4.15–5kb范围内,mir于相距5kb范围内,aluy_old于相距1.5–5kb范围内,alus于相距0.8–5kb范围内及alu_old于相距0.85–5kb范围内;并通过与蒙特卡罗随机模拟(n=100x)22个常染色体的所有cnvts比较,发现所述元件于所述的相距范围内的富集情况达p<0.01。
实施例1-6的测试结果分别显示于图3-8中,在离开各种基因多态性位点,诸如snp1k、mst、shortcnvg、midcnvg、longcnvg和cnvt的近距离内,富集了许多alu、mir和sva元件的插入,经统计学分析,所述富集情况达到p<0.01的水平。这表明在人类基因组进化过程中,逆转录转座子插入所带来的染色体不稳定性促使插入位点附近发生基因型多态性。虽然有些基因型多态性并没有引致个人表型上的差异,另一些却可引致个人表型的差异而被认定为人类性状。图9总结显示,在离开各种snp、mst、cnvg和cnvt位点可被pcr扩增的~3kb近距离内,出现了alu、mir和sva元件的富集情况,这证实了应用alu-、mir-和sva-共有序列基引物进行ite-pcr可以有效地捕获基因组序列进行大规模并行测序(mps),鉴定与人类性状有密切关联的大量基因组多态性位点。
实施例七
由于almivascan测序平台是利用基于alu-、mir-和sva-共有序列设计的ite-pcr引物产生ite-pcr扩增子,并通过mps进行扩增子的序列分析,所以almivascan测序平台的实用性将取决于扩增子的质量。对扩增子质量的要求须具有两个属性,就是高产量及多样性。高产量可提供mps足够的扩增子进行深入测序,而高产量的扩增子,可以通过凝胶电泳和溴化乙锭染色后,在紫外光(uv)照射下它们所呈现的强烈光亮度鉴定;多样性的扩增子可提供大量与alu-、mir-和sva-相邻的广泛序列进行mps分析,而多样性的扩增子可以通过它们在溴化乙锭染色后所呈现的非条带型的弥散状态。本实施例使用了多种h-型和t-型alu-、mir-和sva-序列基引物的组合,应用almivascan测序平台对白血球细胞dna进行分析,并使用扩增子的凝胶电泳,比较不同引物组合所产生的扩增子的产量高低及它们的条带或弥散状态。下面表2描述了所用的不同引物组合。
表2
一组四条alu引物曾被不同研究组报道过了,分别是aluy66h21和aluy278t18(meietal2011,我方研究组),l12a/8(zietkiewiczetal1992)及r12a/267(srivastavaetal2005)。同样地,mir17和mil17(jurkaetal1995)也已报道过了。而aluya5/a8、aluyb8/b9、aluyk4/k12、svah和svat是为本发明应用而设计的引物,是分别基于图六中所显示的aluy子家族的已知序列,该序列富集于midcnvgs的邻近区域,及svas的已知序列设计的(hublyetal2016)。
使用上表2列出的各引物,进行如下所述的ite-pcr:先将15ml全血与30ml内含8.26g氯化铵、1.0g碳酸氢钾、37mgedta的红细胞裂解液温和地混和,并置于4℃孵育30分钟;以3500rpm和4℃冷冻环境离心30分钟分离细胞核;重复裂解和离心步骤直至细胞核沉淀物呈淡黄白色;通过酚/氯仿从沉淀物中提取dna;采用内含特定pcr引物的pcr反应混合物进行ite-pcr扩增。每25μl的反应混合物中内含:2μlbioline10×nh4缓冲液(160mm硫酸铵,670mmtris-hcl,ph8.8,0.1%稳定剂;www.bioline.com),3mm氯化鎂,0.15mmdntp混合物,1单位taq聚合酶,12.5ng白血球细胞dna样品,以及各引物浓度为0.050μm的ite-pcr引物组合。首先,将各反应混合物进行95℃5分钟的dna变性,然后是30个温度循环(一次循环为95℃30秒,50℃30秒和72℃5分钟),最后是72℃7分钟。取8μl反应产物与2μl上样染料混和后,进行琼脂糖凝胶电泳,溴化乙锭染色和uv照射检视。
图10的凝胶电泳染色强度和阵式显示,使用不同引物组合所获得的扩增子dna染色亮度及电泳后模式(呈条带或弥散状图案)均有所不同。泳道c、e和g中的扩增子dna经溴化乙锭染色后呈明显条带状,而泳道b、i、k和m的凝胶染色亮度则相对较弱。由于泳道c和e仅使用mir-序列基引物,泳道g仅使用sva-序列基引物,而泳道b、i、k和m也仅用了少于五种不同的alu-序列基引物,这些结果表明了仅使用mir-序列基引物或sva-序列基引物,又或使用的alu-序列基引物种数不足够,均会产生较低量的扩增子。而泳道d和h除使用了四种不同alu-序列基引物之外,还加上一种mir-序列基或两种sva-序列基引物,结果生成了具充足染色及呈弥散状的扩增子dna;泳道j、l和n各自使用了五种不同的alu-序列基引物,也呈现出强染色弥散模式。值得注意的是,泳道f、o和p所使用的引物组合产生了强烈染色的弥散模式,其组合分别是四种不同alu-序列基引物和两种mir-序列基引物,或七种不同alu-序列基引物与一或两种mir-序列基引物及两种sva-序列基引物。
实施例八
在此实施例中,均采用了实施例七所述泳道a-p的相同引物组合来进行ite-pcr,只是每个25-μlite-pcr反应中用上12.5ng人类痰液dna样品。而获得的凝胶电泳图(见图11)与实施例七的大致相若。泳道c、e和m显现清晰的条带状,泳道f和j则显现颇强染色的弥散模式,而泳道o和p显现强烈染色弥散模式。
实施例九
在此实施例中,均采用了实施例七所述泳道a-p的相同引物组合来进行ite-pcr,只是每个25-μlite-pcr反应中用上125ng人类粪便dna样品。而获得的凝胶电泳图(见图12)与实施例七和八的大致相若。泳道c、i和m为清晰的条带状,泳道j和o则呈现一些强染色的条带,而泳道l和p显现强烈染色弥散模式。
于2011年mei、ding、xue等人发表的科研文章,题为“aluscan:一种应用于扫描人类全基因组序列和结构变异的方法”,详述了使用alu-共有序列基pcr引物扩增alu间序列,所生成的扩增子长度可达6kb,并且电泳图弥散状对扩增子质量可提供了一可靠指标,确定能成功扩增及测序>10mb的人基因组序列。所述序列来自8,000个基因,并在对比一组胶质瘤与对照样品后,发现357个杂合性丢失(lohs)、274个体细胞单核苷酸变异(snvs)、341个体细胞插入/缺失及7个snv热点。实施例7-9采用了不同引物组合进行扩增,电泳图像显示扩增物会随着引物数量和种类递增(见图10-12),这证明在ite-pcr扩增过程中,alu-序列基、mir-序列基和sva-序列基引物之间的协作效应,而相互干扰很少。如图1所示,单一alu-序列基引物仅能扩增基因组alu间序列,但要是合并alu、mir和sva引物一起使用,则能够扩增alu间、mir间、sva间、alu-mir间、alu-sva间和mir-sva间序列,大大扩阔了序列捕获范围。图10-12中,泳道p采用了alu-序列基、mir-序列基的和sva-序列基引物组合,对白血球细胞dna、痰液dna和粪便dna经扩增后,均产生强烈染色和弥散状的扩增产物电泳图。另泳道o、f和l的电泳图亦呈强烈染色和弥散状。综合所述结果,almivascan测序平台能够通过使用不同ite-pcr引物组合,从细胞dna及人体分泌物和排泄物dna中,产生大量不同ite序列片段进行mps分析。图10-12所采用的alu-、mir-和sva-序列基引物组合,仅代表部分而不是全部可能性组合。因此,序列捕获还可通过使用实施例7-9以外的额外alu-、mir-和sva-序列基引物组合进行。同样,almivascan覆盖范围可以扩展至其它逆转录转座子和重复元件的邻近基因组序列,只要在ite-pcr反应混合物中加入基于这些逆转录转座子,和重复元件共有序列设计的pcr引物,条件是该逆转录转座子和重复序列并不出现于微生物dna和病毒dna中。
微生物约占人类粪便干质量达60%(stephenandcummings1980)。在实施例8和9中,用作提取痰液和粪便样品dna的苯酚-氯仿方法,并没有任何去除微生物dna的步骤。因此,可预期这些dna样品将含有各种微生物dna。事实上,由于粪便中的微生物dna含量很高,尽管实施例7和8只用上12.5ng白血球细胞dna或痰液dna来进行ite-pcr反应生成大量人类扩增子,实施例9则必须用上125ng粪便dna才能捕获足够的人类dna扩增子作电泳分析。重要的是,实施例8和9的扩增子电泳图像与实施例7的相类似,证明almivascan分析的应用不受人类粪便中存在的大量微生物dna干扰,甚至其量可能远远超过人类dna量。然而,alu和sva转座子分别仅存于灵长类和人科动物dna中,mir转座子则存于所有哺乳类动物dna中。为了减低人类粪便样品中哺乳动物dna的干扰,因此须于取样前经过72小时无进食任何源自猪肉、牛肉、羊肉或其它哺乳动物肉类或组织,方可进行almivascan分析。若果,almivascan扩增采用了基于l1反转录转座子共有序列的引物,则亦须于取样前72小时内禁食鱼、鸡或其他脊椎动物,以减少鱼或其他脊椎动物插入的l1的干扰,方可进行almivascan分析。图12泳道p和l的凝胶电泳图,就是从所述经过72小时猪、牛、羊肉禁食供样品者粪便dna扩增生成的ite-pcr扩增子,显示强烈染色和弥散模式。这表示虽然样品中存在大量微生物dna,但仍有效地可捕获人类基因组ite-序列。要是没有办法于取样前不进食含有哺乳动物肉类和组织的食品,则必须将mir-共有序列基引物从ite-pcr混合物中剔除,才可进行ite-pcr扩增。
鉴于上述内容,本发明已实现了几个预期目的,并取得有效成果。
由于上述方法可以在不脱离本发明权利范围的情况下作出变更,因此包含在上述说明和附图中的所有内容应被理解为属说明性质而非限制性质的。
美国专利文件:
u.s.5,773,649issuedjune30,1998,entitled"dnamarkerstodetectcancercellsexpressingamutatorphenotypeandmethodofdiagnosisofcancercell"sinnett,etal.
u.s.7,537,889issuedmay26,2009,entitled"assayforquantitationofhumandnausingaluelements"sinhaetal.
u.s.20150225722a1april29,2015,entitled"methodsforselectivetargetingofheterochromatinformingnon-codingrna"f.ozsolek
非专利文献:
bagshawatm,horwoodlj,fergusondmetal(2017).microsatellitepolymorphismsassociatedwithhumanbehavioralandpsychologicalphenotypesincludingagene-environmentinteraction.bmcmedgenet18,12.doi:101186/s12881-017-0374-y
batzerma,deiningerpl(2002).alurepeatsandhumangenomicdiversity.natrevgenet2002,3:370-379.
claussnitzerm,dankeisn,kimkhetal(2015).ftoobesityvariantcircuitryandadiposebrowninginhumans.newengljmed373,895-907.
ding,x.,tsang,sy,xue,hetal(2014).applicationofmachinelearningtodevelopmentofcopynumbervariation-basedpredictionofcancerrisk.genomicsinsights7,1-11.
forbessaetal.(2015).cosmic:exploringtheorld’sknowledgeofsomaticmutationsinhumancancer.nucleicacidsres43,d805-811.
fujimotoa,nishidan,kimuraretal(2009).fgfr2isassociatedwithhairthicknessinasianpopulations.jhumgenet54,461-465.
gianfrancescoo,bubbvj,quinnjp(2017)svaretrotransposonsaspotentialmodulatorsofneuropeptidegeneexpression.neuropeptides64,3-7(2017).
hubleyr,finnrd,clementsjetal.(2016).thedfamdatabaseofrepetitivednafamilies.nucleicacidsres44,d81-89.
iourovly,vorsanovasg,yurovyb(2008).chromosomalmosaicismgoesglobal.molcytogenet1:26.doi10.1186/1755-8166-1-26.
jurkaj,zietkievicze,labudad(1995).ubiquitousmammalian-wideinterspersedrepeats(mirs)aremolecularfossilsfromthemesozoicera.nucleicacidsres23,170-175.
kassdfi,batzerma(1995).inter-alupolymerasechainreaction:advancementsandapplications.analbiochem228,185-193.
kimjj,parkym,baikkhetal.(2012).exomesequencingandsubsequentassociationstudiesidentifyfiveaminoacid-alteringvariantsinfluencinghumanheight.humgenet131,471-478.
krajinovicm,richerc,labudad,sinnettd(1996).detectionofamutatorphenotypeincancercellsbyinter-alupolymerasechainreaction.cancerres56,2733-2737.
kumary,yangj,xuehetal(2015).massiveinsterstitialcopy-neutralloss-of-heterozygosityasevidenceforcancerbeingadiseaseofthedna-damageresponse.bmcmedgenet8:42.doi10.1186/s12920-015-0104-2.
lix,tanl,liuxetal.(2010).agenomewideassociationstudybetweencopynumbervariation(cnv)andhumanheightinchinesepopulation.jgenetgenomics37,779-785.
lows,laucf,xuehetal(2004).associationofsnpsandhaplotypesingabaareceptorβ2genewithschizophrenia.molpsychiatry9,603-608.
mbarekh,steinbergs,nyholtetal(2016).identificationofcommongeneticvariantsinfluencingdizygotictwinningandfemalefertility.amhumgenet98,898-908.
medlandse,nyholtdr.,painteretal(2009).commonvariantsinthetrichohyalingeneareassociatedwithstraighthairineuropeans.amjhumgenet85,750-755.
meil,ding,x,xuehetal.(2011).aluscan:amethodforgenome-widescanningofsequenceandstructurevariationsinthehumangenome.bmcgenomics12:564.doi:10.1186/1471-2164-12-564.
ng,sk,hut,xue,h.etal(2016).featureco-localizationlandscapeofthehumangenome.scirep,6,20650.doi:10.1038/srep20650.
ngsk,xueh(2006).alu-associatedenhancementofsinglenucleotidepolymorphismsinthehumangenome.gene368,110-116.
northbv,curtisd,shampc(2002).anoteonthecalculationofempiricalpvaluesfrommontecarloprocedures.amjhumgenet71,439-441.
payseurba,jingp,haaslrj(2011).agenomicportraitofhumanmicrosatellitevariation.molbiolevol28,303-312.quinnjp,bubbvj(2014)svaretrotransposonsasmodulatorsofgeneexpression.mobilegenetelements4,e32102.
sawayasm,bagshawat,buschiazzoe(2012).promotermicrosatellitesasmodulatorsofhumangeneexpression.intandemrepeatpolymorphisms,ed.hannan,aj.landespp.41-54.
schliena,malkind(2009).copynumbervariationsandcancer.genomicsmed1:62.dof10.1186/gm62.
smitafa,hubleyr,greenp.(2010)repeatmaskeropen-3.0.http://www.repeatmasker.org.1996-2010.
stephenam,cummingsjw(1980).themicrobialcontributiontohumanfecalmass.jmedmicrobiol13,45-56.
srivastavat,setha,dattaketal(2005).inter-alupcrdetectshighfrequencyofgeneticalterationsingliomacellsexposedtosub-lethalcisplatin.internationalj.cancer117:683-9.
sturmra,duffydl,zhaozzetal(2008).asinglesnpinanevolutionaryconservedregionwithinintron86oftheherc2genedetermineshumanblue-browneyecolor.amjhumgenet82,424-431.
walkerja,kilroyge,xingjetal(2003).humandnaquantitationusingaluelement-basedpolymerasechainreaction.analbiochem315,122-128.
zhaoc,xuz,xuehetal(2009).altemative-splicingintheexon-10regionofgabaareceptorβ2subunitgene:relationshipsbetweennovelisoformsandpsychoticdisorders.plosone4,e6977.
zietkiewicze,labudam,sinnettd,glorieuxfh,labudad(1992).linkagemappingbysimultaneousscreeningofmultiplepolymorphiclociusingaluoligonucleotide-directedpcr.procnatlacadsciusa1992,89:8448-8451.
序列表
<110>华晶基因技术有限公司
<120>应用全基因组捕获转座子间区段序列对受微生物污染的人类dna样本
<130>pt20192913-dd-p
<150>us62/507,814
<151>2017-05-18
<160>12
<170>siposequencelisting1.0
<210>1
<211>14
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>consensussequence
<400>1
aaaaatacaaaaaa14
<210>2
<211>21
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>2
tggtctcgatctcctgacctc21
<210>3
<211>18
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>3
gagcgagactccgtctca18
<210>4
<211>12
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>4
tgagccaccgcg12
<210>5
<211>12
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>5
agcgagactccg12
<210>6
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>6
agcttgcagtgagctgagat20
<210>7
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>7
gtccgcagtccggcctgggc20
<210>8
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>8
gatagcgccactgcagtcc19
<210>9
<211>17
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>9
agtgacttgctcaaggt17
<210>10
<211>17
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>10
gcctcagtttcctcatc17
<210>11
<211>19
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>11
agccgagatggcagcagta19
<210>12
<211>20
<212>dna
<213>人工序列(artificialsequence)
<220>
<223>primer
<400>12
accagagacctttgttcact20
- 还没有人留言评论。精彩留言会获得点赞!