一种巴洛沙韦的中间体及其制备方法与应用与流程
本发明涉及医药技术领域,尤其涉及用于制备巴洛沙韦的中间体及其制备方法。
背景技术:
2018年10月24日,瑞士制药巨头罗氏(roche)宣布,美国食品和药物管理局(fda)已批准xofluza(baloxavirmarboxil),该药是一种单剂量、口服药物,用于12岁及以上患者急性、无并发症流感的治疗。此次批准,使xofluza成为近20年来第一种具有新作用机制的流感药物。在临床试验中,xofluza单次治疗即可大幅减少流感症状持续时间,并在仅仅一天内就使病毒排出明显减少。今年2月,baloxavirmarboxil获日本厚生劳动省(mhlw)批准,以品牌名xofluza在日本销售,用于成人及儿科患者a型和b型流感的治疗。
因为患者基数巨大,baloxavir具有非常好的市场潜力。2009年h1n1病毒流行,罗氏的奥司他韦销售额达32亿美元,baloxavir具有更快的抗病毒疗效,服药方案简单,不良反应更小,因此baloxavir非常有望达到奥司他韦的销售额高度。早在2016年,盐野义就将日本与台湾以外地区的开发权转让给了罗氏,baloxavir将成为奥司他韦后时代罗氏继续领跑流感领域的王牌。而在日本制药企业排名10名开外的盐野义也有望通过这一产品进入日本前十,甚至到达前五。
journaloftheamericanchemicalsociety,2006,vol.128,#7,p.2222-2223报道了一种巴洛沙韦的制备方法,其合成路线如下:
以式i化合物为原料,经过苄基保护、甲基氧化、醛基氧化,制得式iv化合物。虽然该路线步骤比较短,但是第二步氧化收率较低,反应温度需要达到155℃,车间生产蒸汽加热很难达到,需要高温油浴的加热方式,氧化过程中使用到剧毒品二氧化硒,二氧化硒是管制化学品,而且对皮肤、粘膜有较强的刺激性,大量吸入其蒸气可引起化学性支气管炎,化学性肺炎或肺水肿,进入眼内可引起结膜炎,可引起接触性皮炎和皮肤灼伤。综上所述,该路线采用剧毒二氧化硒,环境污染严重,不适合工业化生产。
wo2010/11816a1公开了一种巴洛沙韦的制备方法,其合成路线如下:
以式i化合物为原料,经过苄基保护、缩合、磺酰化、消除、双键氧化、醛基氧化,制得式iv化合物。该路线步骤较长,操作繁琐;第三步用到剧毒品甲基磺酰氯,甲基磺酰氯对粘膜、上呼吸道、眼睛和皮肤有强烈刺激性,可致灼伤,吸入后,可因喉和支气管的痉挛、炎症和水肿,化学性肺炎或肺水肿而致死,接触后出现烧灼感、咳嗽、喘息、喉炎、气短、头痛、恶心和呕吐,环境污染严重;第五步氧化用到昂贵的钌催化剂和高碘酸钠联合使用,
成本较高;综上所述,该路线成本较高,且环境污染严重,不适合工业化生产。
鉴于巴洛沙韦良好的药用前景,因此需要开发一种经济、安全的巴洛沙韦的制备方法。
技术实现要素:
本发明涉及一种巴洛沙韦的中间体及其制备方法与应用。本发明提供了一个全新化合物(式iii)及一条制备化合物iv的新路线。该路线以廉价易得的式i化合物作为起始原料,经过苄基保护、缩合反应、氧化反应,制得式iv化合物。
本发明通过巧妙的设计先将甲基间接转变成烯胺,再氧化成醛,可进一步替代甲基的直接氧化,虽然多经过一步中间过程,但是可有效避免采用剧毒的二氧化硒,提高路线的安全性。同时,本发明通过对烯胺氧化条件的选择与优化,采用两种不同的氧化体系,经历两种氧化过程,先将烯胺氧化成醛,醛中间体不经过分离,直接进行下一步反应,可实现“一锅法”将双键氧化到羧酸,有效避免使用昂贵的钌催化剂。本发明设计的整条路线简短新颖,反应条件温和,经济有效,且收率比现有的制备方法高,适于大规模的工业化生产。
本发明涉及一种巴洛沙韦的中间体,其结构如式iii所示:
本发明涉及一种制备中间体iii的方法,式i化合物经过苄基保护、缩合反应得到式iii化合物;
苄基保护试剂为溴苄、氯苄、碘苄,碱为碳酸钾、碳酸钠、氢氧化钠、氢氧化钾、碳酸氢钠、碳酸氢钾、三乙胺、吡啶,溶剂为n,n-二甲基甲酰胺、乙腈、二甲亚砜,式i化合物与苄基保护试剂的摩尔比为1:1~1:2,式i化合物与碱的摩尔比为1:1~1:2,反应温度为45~80℃,反应时间为1~5小时;
缩合试剂为n,n-二甲基甲酰胺二甲缩醛,式ii化合物与n,n-二甲基甲酰胺二甲缩醛的摩尔比为1:1~1:3,溶剂为n,n-二甲基甲酰胺、乙腈、二甲亚砜,反应温度为80~150℃,反应时间为2~10小时。
本申请涉及一种式iv化合物的制备方法,将式iii经过氧化反应得到式iv化合物
其中氧化反应分为两个历程,第一个阶段先将烯胺氧化成醛,氧化剂为高碘酸钠、次氯酸钠、双氧水,式iii化合物与氧化剂的摩尔比为1:1~1:4,反应溶剂为n,n-二甲基甲酰胺、乙腈、二甲亚砜,反应时间为0.5~2小时,反应温度为10~25℃;第二个阶段,将醛基氧化成羧酸,氧化剂体系为亚氯酸钠—双氧水、亚氯酸钠—氨基磺酸,反应溶剂为丙酮和水、乙腈和水,其中丙酮和水是等体积比,乙腈和水是等体积比,反应温度为0~25℃,反应时间为1~2小时。
附图说明。
图1是式ii化合物氢谱图。
图2是式iii化合物氢谱图。
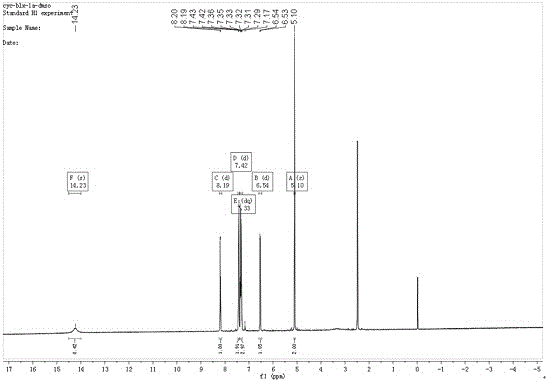
图3是式iv化合物氢谱图。
具体实施方式
以下结合实例说明本发明,但不限制本发明。在本领域内,技术人员对本发明所做的简单替换或改进均属于本发明所保护的技术方案内。
实施例1:
式i化合物经过苄基保护得到式ii化合物
将189g式i化合物溶于1890mln,n-二甲基甲酰胺,加入184ml溴苄,228g碳酸钾,加热至80℃,保温2小时,薄层色谱法检测原料完全转化,降温至室温,加入1000ml四氢呋喃,抽滤除去无机杂质,滤液旋干得到329g白色固体,收率100%。(1hnmr(400mhz,cdcl3)δ7.59(d,j=5.6hz,1h),7.42–7.29(m,5h),6.37(d,j=5.6hz,1h),5.16(s,2h),2.08(s,3h)).
实施例2:
式ii化合物经过缩合反应得到式iii化合物
将54g式ii化合物溶于270mln,n-二甲基甲酰胺,加入74gn,n-二甲基甲酰胺二甲基缩醛,加热至回流,保温8小时,薄层色谱法检测原料完全转化,降温至室温,反应液中加入2000ml水,800ml二氯甲烷萃取3次,合并有机层,无水硫酸钠干燥,浓缩得到55.6g油状物,收率85%。(1hnmr(400mhz,cdcl3)δ7.45(d,j=6.4hz,2h),7.41(d,j=5.6hz,1h),7.36–7.27(m,3h),7.02(d,j=13.4hz,1h),6.21(d,j=5.6hz,1h),5.10(s,2h),5.00(d,j=13.4hz,1h),2.83(s,6h)).
实施例3:
式iii经过氧化反应得到式iv化合物
将73.3g式iii化合物溶于50mln,n-二甲基甲酰胺,加入231g高碘酸钠的100ml水溶液中,控制温度不超过25℃,室温搅拌30min,薄层色谱法检测原料完全转化,将反应液倒入300ml水中,100ml二氯甲烷萃取3次,合并有机层,浓缩得到油状物。将73.3g亚氯酸钠和52.6g氨基磺酸溶于350ml水中,将上述油状物用350ml丙酮溶解,滴加到水溶液中,控制温度不超过25℃,保温1小时,薄层色谱法检测原料完全转化,将反应液浓缩除去丙酮,大量白色固体析出,降温至5℃,抽滤,鼓风干燥,得到54.6g白色固体,收率82%。
1hnmr(400mhz,dmso)δ14.23(s,1h),8.19(d,j=5.6hz,1h),7.42(d,j=6.7hz,2h),7.33(dq,j=14.0,6.8hz,3h),6.54(d,j=5.6hz,1h),5.10(s,2h)).
以上所述的仅是本发明的优选实施方式,应当指出,对于本领域的普通技术人员来说,
在不脱离本发明创造构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!