一种利奈唑胺杂质的合成方法与流程
本发明属于生物医药领域,尤其涉及一种利奈唑胺杂质的合成方法。
背景技术:
利奈唑胺:linezolid,化学名(n-[[(5s)-3-[3-fluoro-4-(4-morpholinyl)phenyl]
-2-oxo-5-oxazolidinyl]methyl]acetamide),商品名为zyvox,是pfizer公司开发研制的第一个噁唑烷酮类抗菌药,2000年在美国上市,用于治疗革兰阳性球菌引起的感染,临床上用于治霉素肠球菌(vre)引起的菌血症,耐甲氧西林金黄色葡萄糖菌(mrsa)引起的肺炎以及耐青霉素肺炎链球菌(prsp)引起的菌血症,复杂性和非复杂性皮肤或皮肤软组织感染、社区获得性肺炎及并发的菌血症等。
利奈唑胺合成过程中,不可避免会生成一些杂质,药物研究人员需要搞清楚原料药开发工艺中杂质种类、来源以及如何控制工艺杂质的产生,为此,就需要开发合成这些杂质的方法,以便得到大量杂质对照品,进而开展利奈唑胺的质量研究。但是关于利奈唑胺杂质的合成方法和路线还未有文献报道。
技术实现要素:
本发明提供了一种利奈唑胺杂质的合成方法,所述合成方法路线合理,可操作性强,产物易提纯,所得的目标产物可用于利奈唑胺检测分析中对杂质进行定性和定量研究,提高利奈唑胺的质量,降低临床用药的风险。
为实现以上目的,本发明采用以下技术方案:一种利奈唑胺杂质的合成方法,包括以下步骤:
(1)将原料ⅰ和boc酸酐溶于溶剂中,加入碱反应,处理色谱柱提纯得到化合物ⅱ;
(2)取步骤(1)制备得到的化合物ⅱ溶于溶剂中,加入还原剂,还原硝基成氨基,处理提纯得到化合物ⅲ;
(3)取步骤(2)得到的化合物ⅲ与化合物ⅳ混合溶于乙醇和水,25℃-100℃反应过夜得到化合物ⅴ;
(4)取步骤(3)得到的化合物ⅴ溶于有机溶剂中,加入碱脱掉邻苯二甲酰亚胺保护基,得到化合物ⅵ;
(5)取步骤(4)得到的化合物ⅵ溶于有机溶剂中,在酸性条件下脱掉boc保护基,在35℃反应2小时,处理纯化得到目标分子化合物ⅶ。
以上所述步骤中,步骤(1)所述的碱为三乙胺、二异丙基乙胺、碳酸氢钠或碳酸钠,反应溶剂为四氢呋喃、二氯甲烷、甲醇或乙醇,在室温条件下反应;原料ⅰ与boc酸酐的摩尔用量比为1:1~1:5,优选1:3;
步骤(2)所述的还原剂为fe/nh4cl、zn/nh4cl或fe/hoac,所述溶剂为乙醇、甲醇或醋酸,加热65℃反应,化合物ⅱ与还原剂的摩尔用量比为1:1~1:5,优选1:3.5;
步骤(3)中化合物ⅲ与化合物ⅳ摩尔用量比为1:1~1:3,优选1:1.5,反应温度为50℃;
步骤(4)所述的碱为甲氨水溶液、水合肼、koh溶液或naoh溶液,所述有机溶剂为甲醇或乙醇,反应温度为25℃-100℃,优选50℃;
步骤(5)所述的酸性条件所述的酸为甲酸、三氟乙酸或盐酸,优选三氟乙酸,所述有机溶剂为二氯甲烷或氯仿。
有益效果:本发明提供了一种利奈唑胺杂质的合成方法,提供一种路线合理,可操作性强,产物易提纯的合成利奈唑胺杂质的方法,该化合物目前未见文献报道。本发明所得的目标产物,可用于利奈唑胺检测分析中对杂质进行定性和定量研究,提高利奈唑胺的质量,降低临床用药的风险。本发明通过大量实验筛选出最优的反应条件和最佳的反应步骤,制备得到的产物对于利奈唑胺合成工艺研究和质量研究具有重要参考价值,对采用外标法来严格控制api质量作出贡献,本发明整个路线设计合理,原料便宜易得,操作简单、所得产品收纯度高,合成的目标分子可作为利奈唑胺检测分析中的杂质标准品,从而加强对该杂质的控制,改进了生产工艺、提高了产品质量。
附图说明
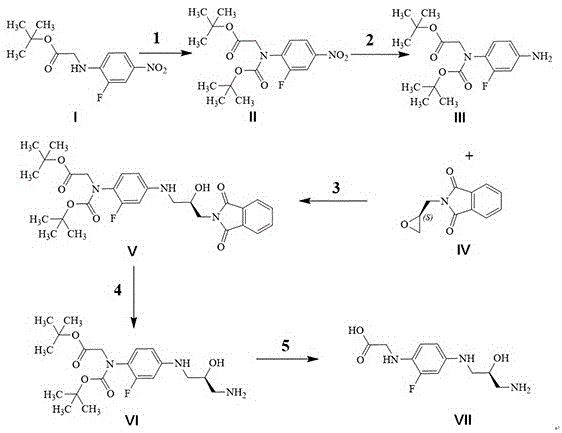
图1为本发明利奈唑胺杂质的合成路线示意图。
具体实施方式
下面结合附图和具体实施例对本发明进行详细说明:
实施例1
如图1所示,一种利奈唑胺杂质的合成方法,包括以下步骤:
步骤(1):化合物ⅱ的制备
6g原料ⅰ和6.7gboc酸酐溶于60ml干燥四氢呋喃中,然后加入5g二异丙基乙胺和0.7g4-二甲氨基吡啶,室温搅拌2小时后,得到一个黄色溶液,tlc显示反应完全,加水淬灭、二氯甲烷萃取,无水硫酸钠干燥后浓缩,用色谱柱提纯得到8g化合物ⅱ,产率97%,ms:369[m-h-];
步骤(2):化合物ⅲ的制备
取8g化合物ⅱ溶于80ml甲醇,加入氯化铵溶液(4g氯化铵溶于10ml水),最后加入5g铁粉,65℃反应8小时,tlc显示反应完全,反应液冷却,浓缩除去甲醇,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到7g化合物ⅲ,产率95%,ms:363[m+na+];
步骤(3):化合物ⅴ的制备
取7g化合物ⅲ溶于150ml乙醇和18ml水,5.1g的化合物ⅳ加入到上述溶液中,反应液于50℃搅拌过夜,点板反应完全,反应液冷却浓缩掉乙醇,水相继续用二氯甲烷萃取两次,硫酸钠干燥后浓缩,用色谱柱提纯得到4.5g化合物ⅴ,产率58%;
步骤(4):化合物ⅵ的制备
取4.5g化合物ⅴ溶于45ml乙醇和45ml40%的koh水溶液,50℃搅拌2小时,浓缩掉乙醇,稀盐酸调ph=6-7,二氯甲烷萃取,有机相用硫酸钠干燥、浓缩得到黄色油状物4g,用色谱柱提纯得到2g化合物ⅵ,产率53%。ms:436[m+na+];
步骤(5):目标产物ⅶ的制备
2g化合物ⅵ溶于50ml二氯甲烷,然后加入5ml的三氟乙酸,35℃反应3小时,薄层色谱显示反应完全,用饱和碳酸钠水溶液中和,有机相蒸干过柱子得到0.85g化合物ⅶ,产率70%。
ms:258[m+h+];or:[α]20d=+25.9°(c=0.4,water);
1hnmr:(400mhz,d2o):δ2.99-3.05(m,1h),3.18-3.22(dd,1h),3.43-3.52(m,2h),4.10(s,1h),4.14-4.16(m,2h),9.76(brs,1h),6.93-7.17(-ar,3h)。
实施例2
步骤(1):化合物ⅱ的制备
3g原料ⅰ和3.3gboc酸酐溶于30ml干燥四氢呋喃中,然后加入2g三乙胺和0.3g4-二甲氨基吡啶,室温搅拌2小时后,得到一个黄色溶液,tlc显示反应完全,加水淬灭、二氯甲烷萃取,用色谱柱提纯得到4g化合物ⅱ,产率97%;
步骤(2):化合物ⅲ的制备
取4g化合物ⅱ溶于40ml甲醇,加入氯化铵溶液(4g氯化铵溶于10ml水),最后加入3g锌粉,65℃反应8小时,tlc显示反应完全,反应液冷却,浓缩除去甲醇,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到3.5g化合物ⅲ,产率95%,ms:363[m+na+];
步骤(3):化合物ⅴ的制备
取3.5g化合物ⅲ溶于75ml乙醇和9ml水,2.6g的化合物ⅳ加入到上述溶液中,反应液于50℃搅拌过夜,点板反应完全,反应液冷却浓缩掉乙醇,水相继续用二氯甲烷萃取两次,硫酸钠干燥后浓缩,用色谱柱提纯得到2.3g化合物ⅴ,产率58%;
步骤(4):化合物ⅵ的制备
取2.3g化合物ⅴ溶于25ml乙醇和25ml水合肼,50℃搅拌2小时,浓缩掉乙醇,稀盐酸调ph=6-7,二氯甲烷萃取,有机相用硫酸钠干燥、浓缩得到黄色油状物3g,用色谱柱提纯得到1g化合物ⅵ,产率53%。ms:436[m+na+];
步骤(5):目标产物ⅶ的制备
1g化合物ⅵ溶于25ml二氯甲烷,然后加入3ml的甲酸,35℃反应3小时,薄层色谱显示反应完全,用饱和碳酸钠水溶液中和,有机相蒸干过柱子得到0.4g化合物ⅶ,产率70%。
实施例3
步骤(1):化合物ⅱ的制备
12g原料ⅰ和14gboc酸酐溶于120ml干燥四氢呋喃中,然后加入10g二异丙基乙胺和1.5g4-二甲氨基吡啶,室温搅拌2小时后,得到一个黄色溶液,tlc显示反应完全,加水淬灭、二氯甲烷萃取,无水硫酸钠干燥后浓缩,用色谱柱提纯得到16.5g化合物ⅱ,产率97.1%;
步骤(2):化合物ⅲ的制备
取16g化合物ⅱ溶于160ml甲醇,加入氯化铵溶液(8g氯化铵溶于20ml水),最后加入10g铁粉,65℃反应8小时,tlc显示反应完全,反应液冷却,浓缩除去甲醇,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到13.5g化合物ⅲ,产率94.5%,ms:363[m+na+];
步骤(3):化合物ⅴ的制备
取14g化合物ⅲ溶于3000ml乙醇和40ml水,10g的化合物ⅳ加入到上述溶液中,反应液于50℃搅拌过夜,点板反应完全,反应液冷却浓缩掉乙醇,水相继续用二氯甲烷萃取两次,硫酸钠干燥后浓缩,用色谱柱提纯得到8.5g化合物ⅴ,产率57.7%;
步骤(4):化合物ⅵ的制备
取9g化合物ⅴ溶于90ml乙醇和90ml40%的甲氨水溶液,50℃搅拌2小时,浓缩掉乙醇,稀盐酸调ph=6-7,二氯甲烷萃取,有机相用硫酸钠干燥、浓缩得到黄色油状物8g,用色谱柱提纯得到4.1g化合物ⅵ,产率53.1%。ms:436[m+na+];
步骤(5):目标产物ⅶ的制备
4g化合物ⅵ溶于80ml甲醇,然后加入5ml的6n浓盐酸,35℃反应3小时,薄层色谱显示反应完全,用饱和碳酸钠水溶液中和,有机相蒸干过柱子得到1.5g化合物ⅶ,产率68%。
实施例4
步骤(1):化合物ⅱ的制备
3g原料ⅰ和3.3gboc酸酐溶于30ml二氯甲烷中,然后加入2g碳酸氢钠和0.3g4-二甲氨基吡啶,室温搅拌2小时后,得到一个黄色溶液,tlc显示反应完全,加水淬灭、二氯甲烷萃取,用色谱柱提纯得到4g化合物ⅱ,产率97%;
步骤(2):化合物ⅲ的制备
取4g化合物ⅱ溶于40ml醋酸,加入hoac溶液,最后加入3g铁粉,65℃反应8小时,tlc显示反应完全,反应液冷却,浓缩除去甲醇,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到3.5g化合物ⅲ,产率95%,ms:363[m+na+];
步骤(3):化合物ⅴ的制备
取3.5g化合物ⅲ溶于75ml乙醇和9ml水,2.6g的化合物ⅳ加入到上述溶液中,反应液于50℃搅拌过夜,点板反应完全,反应液冷却浓缩掉乙醇,水相继续用二氯甲烷萃取两次,硫酸钠干燥后浓缩,用色谱柱提纯得到2.3g化合物ⅴ,产率58%;
步骤(4):化合物ⅵ的制备
取2.3g化合物ⅴ溶于25ml乙醇和25ml氢氧化钠溶液,25℃搅拌2小时,浓缩掉乙醇,稀盐酸调ph=6-7,二氯甲烷萃取,有机相用硫酸钠干燥、浓缩得到黄色油状物3g,用色谱柱提纯得到1g化合物ⅵ,产率53%。ms:436[m+na+];
步骤(5):目标产物ⅶ的制备
1g化合物ⅵ溶于25ml二氯甲烷,然后加入3ml的甲酸,35℃反应3小时,薄层色谱显示反应完全,用饱和碳酸钠水溶液中和,有机相蒸干过柱子得到0.4g化合物ⅶ,产率70%。
实施例5
步骤(1):化合物ⅱ的制备
12g原料ⅰ和14gboc酸酐溶于120ml干燥二氯甲烷中,然后加入10g碳酸氢钠和8g碳酸钠,室温搅拌2小时后,得到一个黄色溶液,tlc显示反应完全,加水淬灭、二氯甲烷萃取,无水硫酸钠干燥后浓缩,用色谱柱提纯得到16.5g化合物ⅱ,产率96%;
步骤(2):化合物ⅲ的制备
取16g化合物ⅱ溶于160ml甲醇,加入氯化铵溶液(8g氯化铵溶于20ml水),最后加入10g铁粉,65℃反应8小时,tlc显示反应完全,反应液冷却,浓缩除去甲醇,水相用乙酸乙酯萃取,无水硫酸钠干燥,浓缩得到13.5g化合物ⅲ,产率94.5%,ms:363[m+na+];
步骤(3):化合物ⅴ的制备
取14g化合物ⅲ溶于3000ml乙醇和40ml水,10g的化合物ⅳ加入到上述溶液中,反应液于50℃搅拌过夜,点板反应完全,反应液冷却浓缩掉乙醇,水相继续用二氯甲烷萃取两次,硫酸钠干燥后浓缩,用色谱柱提纯得到8.5g化合物ⅴ,产率57.7%;
步骤(4):化合物ⅵ的制备
取9g化合物ⅴ溶于90ml乙醇和90ml40%的水合肼,100℃搅拌2小时,浓缩掉乙醇,稀盐酸调ph=6-7,二氯甲烷萃取,有机相用硫酸钠干燥、浓缩得到黄色油状物8g,用色谱柱提纯得到4.1g化合物ⅵ,产率53.1%。ms:436[m+na+];
步骤(5):目标产物ⅶ的制备
4g化合物ⅵ溶于80ml氯仿,然后加入5ml的6n浓盐酸,35℃反应3小时,薄层色谱显示反应完全,用饱和碳酸钠水溶液中和,有机相蒸干过柱子得到1.5g化合物ⅶ,产率68%。
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
- 还没有人留言评论。精彩留言会获得点赞!