具有杀菌消毒活性的哒嗪类化合物及其制备方法和应用与流程
本发明属于抗菌药物合成技术领域,具体涉及一种具有杀菌消毒活性的哒嗪类化合物及其制备方法和应用。
背景技术:
哒嗪又称邻二氮杂苯,是一类具有特殊结构且具有广泛生物活性的含氮杂环化合物。自从哒嗪类除草剂maleichy-droazide被成功开发后,哒嗪类化合物的研究得到了快速的发展。例如哒嗪霉菌素是第一例具有天然杀菌活性的哒嗪化合物。研究发现哒嗪化合物在医药方面存在较好的生物活性,目前已经有多种哒嗪类药物开发上市,如治疗心理疾病的药物米那普林、抗菌药物长效磺胺、降压药物双肼苯哒嗪、广谱抗生素类药物磺胺氯哒嗪促进血液循环药物甲硫阿美铵等。
目前,关于哒嗪化合物的合成文献报道较多,哒嗪化合物的合成路线主要有以下几种:1、以丁烯二醛为原料合成:由丁烯二醛和水合肼经过diels-alder反应合成。此方法来合成步骤少,但是反应条件较难控制,易发生链式反应,而导致收率较低;2、以甲脒乙酸盐为原料合成:用乙醇和原甲酸三乙酯在加热的条件下加入氨气生成,得到产物甲脒乙酸盐。水合肼是在醋酸等条件下反应生成四嗪,再通过和n,n-二甲基乙烯胺经diels-alder反应,最终合成产物;该方法的产率仅为9.7%。得出的中间产物四嗪是挥发性的,它的分离和保存也较为困难;3、顺丁烯二酸酐与水合肼反应生成:通过顺丁烯二酸酐与水合肼反应生成顺丁烯二酰肼,再用三氯氧磷氯化生成3,6-二氯哒嗪,最后以氨水为缚酸剂,经pd/c催化剂催化加氢最终合成,产品收率为42%;4、以哒嗪母核为原料的合成:例如合成甲基哒嗪,是以哒嗪为原料,和醋酸或叔丁醇在浓硫酸和其他氧化剂的作用下生成甲基哒嗪。
鉴于哒嗪化合物是一类具有特殊结构且具有广泛生物活性的含氮杂环化合物,该类化合物的研究和应用对与农药和医药领域的发展具有重要的作用。因此,开发具有新型结构的哒嗪类化合物分子,以及开发新型哒嗪类化合物的制备方法,扩展哒嗪类化合物的用途的同时降低生产成本具有很重要的现实意义。
技术实现要素:
本发明解决的技术问题是提供了一种具有杀菌消毒活性的哒嗪类化合物及其制备方法和应用。
本发明为解决上述技术问题采用如下技术方案,一种具有杀菌消毒活性的哒嗪类化合物的结构为:
本发明为解决上述技术问题采用如下技术方案,一种具有杀菌消毒活性的哒嗪类化合物的制备方法,其特征在于具体步骤为:
(1)3-(甲胺基)-4h-1,2,4-三氮唑-4-胺的合成;
(2)3-(甲胺基)-4h-1,2,4-三氮唑-4-胺与4-甲基苯乙酮发生缩合反应得到3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺;
(3)3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺经缩合后成环得到(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺;
(4)(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺与氯乙酰氯反应得到氯代化合物;
(5)氯代化合物与4-吡啶硫醇反应得到目标化合物。
步骤(1)为采用以下两种方法中的一种:
a、把一定量的盐酸肼加入水,搅拌均匀后向反应液中加入盐酸胍,在室温条件下向反应液中加入浓度为80%的水合肼,然后升温至回流,反应过程中有刺鼻性气体放出,通过观察反应瓶中的气泡逐渐减少消失后停止加热,冷却至室温,向反应液中滴加稀盐酸调节反应液ph到2~3,搅拌过程中有晶体析出,抽滤得到固体和一定量的甘氨酸和碳酸钾加入甲苯中,加热至回流,反应结束后在真空条件下除去部分甲苯,然后向反应体系中加入水,在10℃条件下搅拌至有固体析出,抽滤得到3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺;把所得到的3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺加入水中,再缓慢滴加一定量的过氧化物,滴加完后缓慢加热至回流;反应结束后加入活性炭,搅拌一段时间脱色,趁热过滤,用乙酸乙酯萃取反应液多次,合并有机相,再用无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺;所述的盐酸胍与盐酸肼的投料量摩尔比为1:0.5;所述的盐酸胍与甘氨酸与碳酸钾的投料量摩尔比为1:1~1.2:1;所述的过氧化物为双氧水、过氧乙酸或过氧三氟乙酸;所述的氧化物为过氧乙酸或过氧三氟乙酸时,盐酸胍与过氧化物的投料量摩尔比为1:2;
b、把一定量的2-氨基乙酰亚氨酸乙酯和甲酰肼加入四氢呋喃中,在氩气保护下,缓慢加热至回流,通过分水器除去反应体系中的水分,浓缩反应液,然后加入一定量的含量为80%的水合肼和四氢呋喃,继续加热至回流,通过分水器除去反应体系中产生的水,反应一段时间后向反应液中加入活性炭,搅拌后趁热抽滤,然后加入水,搅拌一段时间,真空条件下蒸除四氢呋喃,再用乙酸乙酯萃取有机相多次,合并有机相,经无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺;所述的2-氨基乙酰亚氨酸乙酯与甲酰肼的投料量摩尔比为1:1.5~2.5;所述的2-氨基乙酰亚氨酸乙酯与含量为80%的水合肼的投料量质量比为1:10。
步骤(2)为:把一定量的3-(甲胺基)-4h-1,2,4-三氮唑-4-胺)和4-甲基苯乙酮加入到甲苯中,在室温条件下,搅拌均匀后加入氢氧化钡,然后逐渐升温至回流,及时除去反应过程中生成的水,反应结束后倒入水中,再用稀盐酸调节反应液ph为中性,然后用二氯甲烷萃取反应液多次,合并有机相,再经无水硫酸镁干燥后浓缩得到3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺;所述的3-(甲胺基)-4h-1,2,4-三氮唑-4-胺与4-甲基苯乙酮与氢氧化钡的投料量摩尔比为1:1:1。
步骤(3)为:把一定量的3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺和叔丁醇钾加入到n,n-二甲基甲酰胺中,在室温条件下搅拌均匀,加入溶有一定量叔丁氧基二(二甲基氨基)甲烷的n,n-二甲基甲酰胺溶液,继续在室温条件反应至原料反应完全后,向反应液中加入水,再用稀盐酸溶液调节反应液ph至5~6,然后用二氯甲烷萃取反应液多次,合并有机相,浓缩后向反应液中加入有机酸,滴加完后搅拌均匀,发现溶液呈现黄色浑浊,缓慢升高温度,当温度达到70℃时,会发现溶液呈现澄清状态,在该温度条件下反应一段时间,向反应液中加水,然后再用饱和碳酸氢钠溶液调节反应液ph为中性,再用二氯甲烷萃取反应液多次,合并有机相,经无水硫酸镁干燥后浓缩后经硅胶柱层析分离得到(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺;所述的3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺与叔丁醇钾与叔丁氧基二(二甲基氨基)甲烷的投料量摩尔比为1:1~1.2:1~1.2;所述的有机酸为冰乙酸或三氟乙酸。
步骤(4)为:把一定量的(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺加入到干燥的二氯甲烷中搅拌溶解,将体系置于10℃条件下逐滴加入溶有氯乙酰氯的二氯甲烷溶液,滴加完毕后,逐渐恢复至室温,反应结束后加入饱和碳酸氢钠溶液调节反应液ph为中性,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩得到氯代化合物;所述的(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺与氯乙酰氯的投料量摩尔比为1:1.2。
步骤(5)为:称取一定量的氯代化合物和碱性化合物加入到瓶中,在氮气保护下,再加入干燥的二氯甲烷搅拌均匀,将体系置于10℃条件下逐滴加入溶有4-吡啶硫醇的二氯甲烷溶液,滴加完毕后,逐渐恢复至室温,反应结束后向反应液中加入饱和碳酸钠溶液调节反应液ph,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩,然后经硅胶柱层析分离提纯得到目标化合物;所述的碱性化合物为甲醇钠或碳酸钾;所述的氯代化合物与碱性化合物与4-吡啶硫醇的投料量摩尔比为1:1~2:1~1.2。
本发明具有的有益效果:本发明通过新方法合成了一种结构新颖的哒嗪类化合物,并通过微量二倍稀释法进行抗菌活性测试,发现目标化合物对金黄色葡萄球菌具有良好的抗菌作用。
附图说明
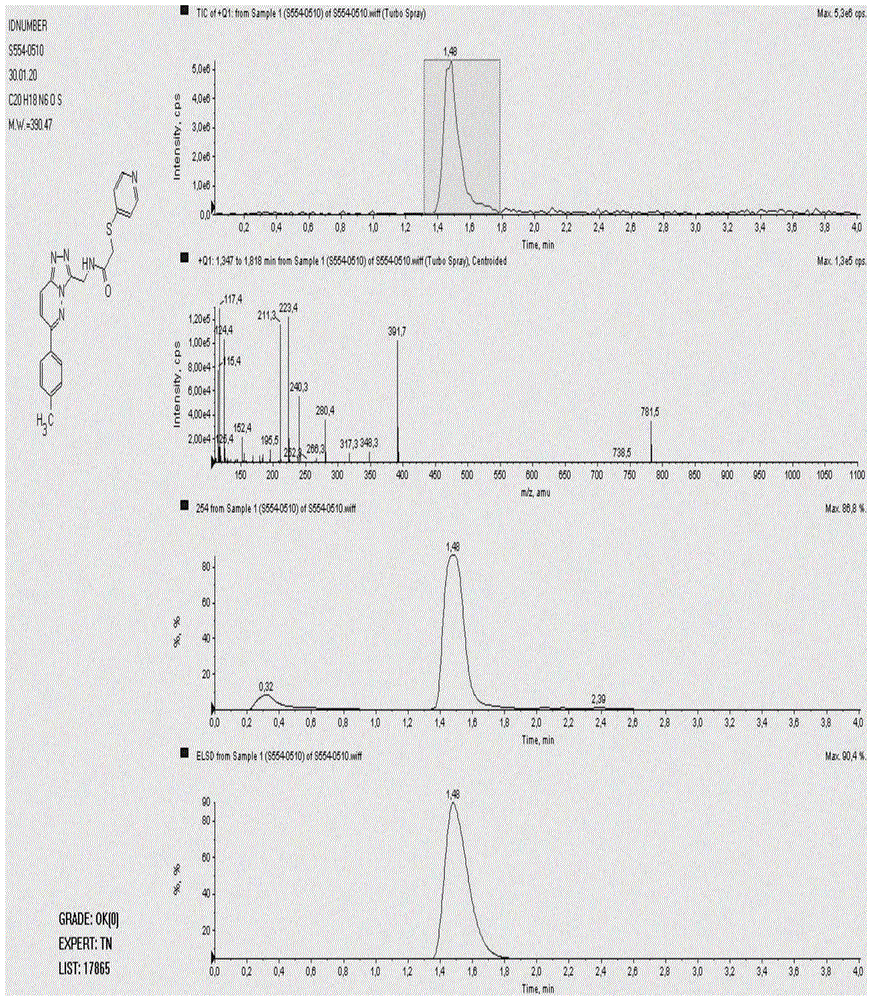
图1是目标化合物的质谱图。
具体实施方式
以下通过实施例对本发明的上述内容做进一步详细说明,但不应该将此理解为本发明上述主题的范围仅限于以下的实施例,凡基于本发明上述内容实现的技术均属于本发明的范围。
实施例1
在带有冷凝器的多功能反应瓶中,把盐酸肼5g加入水50ml,搅拌均匀后向反应液中加入盐酸胍9.5g,在室温条件下搅拌30min,然后向反应液中加入浓度为80%的水合肼100g,然后升温至回流,回流过程中有刺鼻性气体放出,通过观察大约回流反应20min后,反应瓶中的气泡逐渐减少消失,然后继续回流反应20min,停止加热,冷却至室温,向反应液中滴加稀盐酸调节反应液ph到2~3,搅拌过程中有晶体析出,抽滤得到固体和甘氨酸9g加入甲苯70ml中,搅拌均匀后加入碳酸钾14g,然后加热至回流,反应2h,在真空条件下除去甲苯30ml,然后向反应体系中加入水100ml,在10℃条件下搅拌10min,有固体析出,抽滤得到中间体3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺;把所得到的中间体加入到水150ml中,再缓慢滴加含量为20%的双氧水100ml,滴加完后缓慢加热至回流;反应5h,通过tlc监控(需要碘熏)原料反应完全后,向反应液中加入活性炭2g,搅拌20min,趁热过滤,用乙酸乙酯50ml萃取反应液多次,合并有机相,再用无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺7.5g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例2
在带有冷凝器的多功能反应瓶中,把盐酸肼5g加入水50ml,搅拌均匀后向反应液中加入盐酸胍9.5g,在室温条件下搅拌30min,然后向反应液中加入浓度为80%的水合肼100g,然后升温至回流,回流过程中有刺鼻性气体放出,通过观察大约回流反应20min后,反应瓶中的气泡逐渐减少消失,然后继续回流反应20min,停止加热,冷却至室温,向反应液中滴加稀盐酸调节反应液ph到2~3,搅拌过程中有晶体析出,抽滤得到固体和甘氨酸9g加入甲苯70ml中,搅拌均匀后加入碳酸钾14g,然后加热至回流,反应2h,在真空条件下除去甲苯30ml,然后向反应体系中加入水100ml,在10℃条件下搅拌10min,有固体析出,抽滤得到中间体3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺;把所得到的中间体和过氧乙酸15g加入到水100ml中,搅拌均匀后缓慢加热至回流;反应3h,通过tlc监控(需要碘熏)原料反应完全后,向反应液中加入活性炭2g,搅拌20min,趁热过滤,用乙酸乙酯50ml萃取反应液多次,合并有机相,再用无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺9.2g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例3
在带有冷凝器的多功能反应瓶中,把盐酸肼5g加入水50ml,搅拌均匀后向反应液中加入盐酸胍9.5g,在室温条件下搅拌30min,然后向反应液中加入浓度为80%的水合肼100g,然后升温至回流,回流过程中有刺鼻性气体放出,通过观察大约回流反应20min后,反应瓶中的气泡逐渐减少消失,然后继续回流反应20min,停止加热,冷却至室温,向反应液中滴加稀盐酸调节反应液ph到2~3,搅拌过程中有晶体析出,抽滤得到固体和甘氨酸9g加入甲苯70ml中,搅拌均匀后加入碳酸钾14g,然后加热至回流,反应2h,在真空条件下除去甲苯30ml,然后向反应体系中加入水100ml,在10℃条件下搅拌10min,有固体析出,抽滤得到中间体3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺;把所得到的中间体和过氧三氟乙酸20g(0.15mol)加入到水中,搅拌均匀后缓慢加热至回流;反应5h,通过tlc监控(需要碘熏)原料反应完全后,向反应液中加入活性炭2g,搅拌20min,趁热过滤,用乙酸乙酯50ml萃取反应液多次,合并有机相,再用无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺9.7g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例4
在带有冷凝器的多功能反应瓶中,把盐酸肼5g加入水50ml,搅拌均匀后向反应液中加入盐酸胍9.5g,在室温条件下搅拌30min,然后向反应液中加入浓度为80%的水合肼100g,然后升温至回流,回流过程中有刺鼻性气体放出,通过观察大约回流反应20min后,反应瓶中的气泡逐渐减少消失,然后继续回流反应20min,停止加热,冷却至室温,向反应液中滴加稀盐酸调节反应液ph到2~3,搅拌过程中有晶体析出,抽滤得到固体和甘氨酸9g加入甲苯70ml中,搅拌均匀后加入碳酸钾14g,然后加热至回流,反应2h,在真空条件下除去甲苯30ml,然后向反应体系中加入水100ml,在10℃条件下搅拌10min,有固体析出,抽滤得到3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺;把所得到的3-(甲胺基)-5-肼基-4h-1,2,4-三氮唑-4-胺和无水硫酸铜16g加入到水100ml和叔丁醇20ml的混合溶液中,缓慢加热至回流,反应2.5h,搅拌反应过程中有大量气泡冒出,应该为反应生成的氮气,通过tlc监控原料反应完全后,向反应液中加入活性炭3g,搅拌20min,趁热过滤,滤液用乙酸乙酯50ml萃取反应液多次,合并有机相,再用无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺8.8g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例5
在带有分水器的500ml反应瓶中,加入四氢呋喃180ml,再加入2-氨基乙酰亚氨酸乙酯10g和甲酰肼12g,在氩气保护下,缓慢加热至回流,通过分水器除去反应体系中的水分,经过50min,浓缩反应液,然后加入含量为80%的水合肼100g和四氢呋喃200ml,继续加热至回流,通过分水器及时除去反应体系中产生的水,回流反应6.5h后向反应液中加入活性炭2g,搅拌10min后趁热抽滤,然后加入水100ml,搅拌10min,真空条件下整除四氢呋喃,再用乙酸乙酯萃取有机相多次,合并有机相,经无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺10.1g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例6
在带有分水器的500ml反应瓶中,加入四氢呋喃180ml,再加入2-氨基乙酰亚氨酸乙酯10g和甲酰肼9g,在氩气保护下,缓慢加热至回流,通过分水器除去反应体系中的水分,经过50min,浓缩反应液,然后加入含量为80%的水合肼100g和四氢呋喃200ml,继续加热至回流,通过分水器及时除去反应体系中产生的水,回流反应6.5h后向反应液中加入活性炭2g,搅拌10min后趁热抽滤,然后加入水100ml,搅拌10min,真空条件下整除四氢呋喃,再用乙酸乙酯萃取有机相多次,合并有机相,经无水硫酸镁干燥后浓缩,再经硅胶柱层析分离得到3-(甲胺基)-4h-1,2,4-三氮唑-4-胺6.5g;1hnmr(400mhz,dmso-d6):8.87(s,2h),8.55(s,1h),5.41(s,2h),4.67(s,2h);元素分析计算值[c3h7n5]:c,31.85;h,6.24;n,61.91.实测值:c,31.93;h,6.28;n,61.79。
实施例7
在反应瓶中,把3-(甲胺基)-4h-1,2,4-三氮唑-4-胺11.5g和4-甲基苯乙酮13.5g加入到甲苯200ml中,在室温条件下,搅拌均匀后加入氢氧化钡17g,然后逐渐升温至回流,及时除去反应过程中生成的水,大约回流反应3h,tlc监控原料反应完全后,向反应液中倒入水200ml中,用稀盐酸调节反应液ph为中性,然后用二氯甲烷50ml萃取反应液多次,合并有机相,再经无水硫酸镁干燥后浓缩得到3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺18.5g;ms(esi)m/z:230.3(m+h+)。
实施例8
在反应瓶中,把3-(甲胺基)-4h-1,2,4-三氮唑-4-胺11.5g和4-甲基苯乙酮16g加入到甲苯200ml中,在室温条件下,搅拌均匀后加入氢氧化钡17g(0.1mol),然后逐渐升温至回流,及时除去反应过程中生成的水,大约回流反应2h,tlc监控原料反应完全后,向反应液中倒入水200ml中,用稀盐酸调节反应液ph为中性,然后用二氯甲烷50ml萃取反应液多次,合并有机相,再经无水硫酸镁干燥后浓缩得到3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺19.1g;ms(esi)m/z:230.3(m+h+)。
实施例9
在反应瓶中,把3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺23g和叔丁醇钾11.5g加入到n,n-二甲基甲酰胺200ml,在室温条件下搅拌均匀,加入溶有叔丁氧基二(二甲基氨基)甲烷18g的n,n-二甲基甲酰胺溶液50ml,继续在室温条件反应3.5h,tlc监控原料反应完全后,向反应液中加入水150ml,再用稀盐酸溶液调节反应液ph至5~6,然后用二氯甲烷萃取反应液多次,合并有机相,浓缩后向反应液中加入冰乙酸100ml,滴加完后搅拌均匀,发现溶液呈现黄色浑浊,缓慢升高温度,当温度达到70℃时,会发现溶液呈现澄清状态,在该温度条件下反应2h,减压蒸除冰乙酸50ml,向反应液中加水200ml,然后再用饱和碳酸氢钠溶液调节反应液ph为中性,再用二氯甲烷萃取反应液多次,合并有机相,经无水硫酸镁干燥后浓缩后经硅胶柱层析分离得到(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺14.9g;1hnmr(400mhz,cdcl3):8.51(d,j=4.0hz,1h),8.37(s,2h),8.14(d,j=4.0hz,1h),7.73(d,j=8.0hz,2h),7.35(d,j=8.0hz,2h),2.92(s,2h),2.28(s,3h)。
实施例10
在反应瓶中,把3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺23g和叔丁醇钾11.5g加入到n,n-二甲基甲酰胺200ml中,在室温条件下搅拌均匀,加入溶有叔丁氧基二(二甲基氨基)甲烷18g的n,n-二甲基甲酰胺溶液50ml,继续在室温条件反应3.5h,tlc监控原料反应完全后,向反应液中加入水150ml,再用稀盐酸溶液调节反应液ph至5~6,然后用二氯甲烷萃取反应液多次,合并有机相,浓缩后向反应液中加入三氟乙酸80ml,滴加完后搅拌均匀,发现溶液呈现黄色浑浊,缓慢升高温度,当温度达到50℃时,会发现溶液呈现澄清状态,在该温度条件下反应2h,向反应液中加水200ml,然后再用饱和碳酸氢钠溶液调节反应液ph为中性,再用二氯甲烷萃取反应液多次,合并有机相,经无水硫酸镁干燥后浓缩经硅胶柱层析分离得到(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺19.7g;1hnmr(400mhz,cdcl3):8.51(d,j=4.0hz,1h),8.37(s,2h),8.14(d,j=4.0hz,1h),7.73(d,j=8.0hz,2h),7.35(d,j=8.0hz,2h),2.92(s,2h),2.28(s,3h)。
实施例11
在反应瓶中,把3-氨甲基-h-(1-(4-甲苯基)-亚乙基)-4h-1,2,4-三氮唑-4-胺23g和叔丁醇钾11.5g加入到n,n-二甲基甲酰胺200ml中,在室温条件下搅拌均匀,加入溶有叔丁氧基二(二甲基氨基)甲烷27g的n,n-二甲基甲酰胺溶液50ml,继续在室温条件反应3.5h,tlc监控原料反应完全后,向反应液中加入水150ml,再用稀盐酸溶液调节反应液ph至5~6,然后用二氯甲烷萃取反应液多次,合并有机相,浓缩后向反应液中加入三氟乙酸80ml,滴加完后搅拌均匀,发现溶液呈现黄色浑浊,缓慢升高温度,当温度达到50℃时,会发现溶液呈现澄清状态,在该温度条件下反应2h,向反应液中加水200ml,然后再用饱和碳酸氢钠溶液调节反应液ph为中性,再用二氯甲烷萃取反应液多次,合并有机相,经无水硫酸镁干燥后浓缩经硅胶柱层析分离得到(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺20.3g;1hnmr(400mhz,cdcl3):8.51(d,j=4.0hz,1h),8.37(s,2h),8.14(d,j=4.0hz,1h),7.73(d,j=8.0hz,2h),7.35(d,j=8.0hz,2h),2.92(s,2h),2.28(s,3h)。
实施例12
在带有搅拌的多口反应瓶中,称(6-(4-苯基)-1,2,4-三氮唑[4,3-b]哒嗪-3-基)甲基胺24g加入到干燥的二氯甲烷180ml中搅拌溶解,将体系置于10℃条件下逐滴加入溶有氯乙酰氯14g的二氯甲烷溶液50ml,滴加完毕后,逐渐恢复至室温,tlc检测反应结束,向反应液中加入饱和碳酸氢钠溶液120ml,调节反应液ph为中性,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩得到氯代化合物27.5g;ms(esi)m/z:316.9(m+h+)。
实施例13
在带有搅拌的多口反应瓶中,称氯代化合物32g和甲醇钠11g加入到瓶中,氮气置换三次,在氮气保护下,再加入干燥的二氯甲烷250ml搅拌均匀,将体系置于10℃条件下逐滴加入溶有4-吡啶硫醇13.5g的二氯甲烷溶液50ml,滴加完毕后,逐渐恢复至室温,tlc检测反应结束,向反应液中加入饱和碳酸钠溶液75ml,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩,然后经硅胶柱层析分离提纯得到目标化合物36.4g;ms(esi)m/z:391.7(m+h+);1hnmr(400mhz,dmso-d6):8.93(d,j=8.0hz,2h),8.37-8.35(m,1h),8.07(d,j=4.0hz,1h),8.02(s,1h),7.73(d,j=8.0hz,2h),7.38-7.35(m,2h),7.11(d,j=4.0hz,2h),4.52(s,2h),3.92(s,2h),2.31(s,3h);元素分析计算值[c20h18n6os]:c,61.52;h,4.65;n,21.52.实测值:c,61.29;h,4.68;n,21.37。
实施例14
在带有搅拌的多口反应瓶中,称氯代化合物32g和碳酸钾27g加入到瓶中,氮气置换三次,在氮气保护下,再加入干燥的二氯甲烷250ml搅拌均匀,将体系置于10℃条件下逐滴加入溶有4-吡啶硫醇13.5g的二氯甲烷溶液50ml,滴加完毕后,逐渐恢复至室温,tlc检测反应结束,向反应液中加入饱和碳酸钠溶液75ml,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩,然后经硅胶柱层析分离提纯得到目标化合物30.7g;ms(esi)m/z:391.7(m+h+);1hnmr(400mhz,dmso-d6):8.93(d,j=8.0hz,2h),8.37-8.35(m,1h),8.07(d,j=4.0hz,1h),8.02(s,1h),7.73(d,j=8.0hz,2h),7.38-7.35(m,2h),7.11(d,j=4.0hz,2h),4.52(s,2h),3.92(s,2h),2.31(s,3h);元素分析计算值[c20h18n6os]:c,61.52;h,4.65;n,21.52.实测值:c,61.29;h,4.68;n,21.37。
实施例15
在带有搅拌的多口反应瓶中,称氯代化合物32g和碳酸钾13.5g加入到瓶中,氮气置换三次,在氮气保护下,再加入干燥的二氯甲烷250ml搅拌均匀,将体系置于10℃条件下逐滴加入溶有4-吡啶硫醇13.5g的二氯甲烷溶液50ml,滴加完毕后,逐渐恢复至室温,tlc检测反应结束,向反应液中加入饱和碳酸钠溶液75ml,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩,然后经硅胶柱层析分离提纯得到目标化合物22.9g;ms(esi)m/z:391.7(m+h+);1hnmr(400mhz,dmso-d6):8.93(d,j=8.0hz,2h),8.37-8.35(m,1h),8.07(d,j=4.0hz,1h),8.02(s,1h),7.73(d,j=8.0hz,2h),7.38-7.35(m,2h),7.11(d,j=4.0hz,2h),4.52(s,2h),3.92(s,2h),2.31(s,3h);元素分析计算值[c20h18n6os]:c,61.52;h,4.65;n,21.52.实测值:c,61.29;h,4.68;n,21.37。
实施例16
在带有搅拌的多口反应瓶中,称氯代化合物32g和碳酸钾13.5g加入到瓶中,氮气置换三次,在氮气保护下,再加入干燥的二氯甲烷250ml搅拌均匀,将体系置于10℃条件下逐滴加入溶有4-吡啶硫醇11g的二氯甲烷溶液50ml,滴加完毕后,逐渐恢复至室温,tlc检测反应结束,向反应液中加入饱和碳酸钠溶液75ml,然后分出有机相,水相用二氯甲烷进行萃取,合并有机相后进行浓缩,然后经硅胶柱层析分离提纯得到目标化合物17.2g;ms(esi)m/z:391.7(m+h+);1hnmr(400mhz,dmso-d6):8.93(d,j=8.0hz,2h),8.37-8.35(m,1h),8.07(d,j=4.0hz,1h),8.02(s,1h),7.73(d,j=8.0hz,2h),7.38-7.35(m,2h),7.11(d,j=4.0hz,2h),4.52(s,2h),3.92(s,2h),2.31(s,3h);元素分析计算值[c20h18n6os]:c,61.52;h,4.65;n,21.52.实测值:c,61.29;h,4.68;n,21.37。
实施例17
采用微量二倍稀释法测硫酸链霉素和目标化合物对革兰氏阴性菌(大肠杆菌)和革兰氏阳性菌(金色葡萄球菌)的抗菌活性。
lb液体培养基配制:称取胰蛋白胨10.0g、酵母提取物5.0g、氯化钠10.0g溶解于蒸馏水中,并定容至1l。在室温下用稀hcl(1mol/l)或稀naoh(1mol/l)调ph为7.0±0.1。121℃高压灭菌15min,备用。
在无菌的96孔板中每孔加入100μl的lb液体培养液,三排为一组,在第一个孔加入待测化合物母液100μl,然后对药物进行二倍稀释。即第一孔中加入药液后用移液枪充分吹打(至少三次以上)使药物与lb液体培养基充分混匀,然后吸取100μl加入第二孔再充分吹打使之与lb液体培养基充分混匀,照此重复至第十个孔,第10列吸出100μl扔掉。再在每一孔加入稀释好的菌液100μl。在同一块板的第11列做一列阴性对照(仅加空白lb液体培养基不加菌液)和在第12列上做一列阳性对照(加菌液不加药液)。目标化合物及对照药物按上述方法依次加药。各个药物的终浓度分别为128、64、32、16、8、4、2、1、0.5、0.25μmol/ml。置于37℃恒温振荡培养箱中培养14h、16h、20h,观察结果,每个样品做3个重复。根据观察的结果,若有细菌生长则孔板底部会出现白色沉淀,再用上述方法进行进一步的浓度筛选,最终以未有沉淀的最小药物浓度作为mic值。最终测得目标化合物对大肠杆菌和金色葡萄球菌的最小药物浓度分别为mic为32μmol/ml和0.5μmol/ml。
以上实施例描述了本发明的基本原理、主要特征及优点,本行业的技术人员应该了解,本发明不受上述实施例的限制,上述实施例和说明书中描述的只是说明本发明的原理,在不脱离本发明原理的范围下,本发明还会有各种变化和改进,这些变化和改进均落入本发明保护的范围内。
- 还没有人留言评论。精彩留言会获得点赞!