一种制备格列美脲晶型Ⅰ的方法与流程
本发明涉及一种格列美脲晶型i的制备方法,属于原料药制备技术领域。
背景技术:
格列美脲(式i化合物)作为一种新型的磺胺类降糖药物,用于2型糖尿病的治疗。国外研究发现其具有较强的膜外降糖作用,包括增加胰岛素敏感性及模拟外周膜岛素作用,改善葡萄糖转运子(glut-4)的转位/去磷酸化,增加葡萄糖在外周组织的摄取。在所有的磺酰胺类药物中,格列美脲的血浆胰岛素升高与血糖降低比值是最低的。
格列美脲常见的存在形式有:无定型、晶型i、晶型ⅱ、国内有α、β、γ、δ、ε等晶型的报道。其中,晶型i为药用晶型,是相对较稳定的晶型,其它晶型未见作为活性成分的报道。晶型i的制备方法的公开文献并不多。
最早公开的晶型i制备方法(iwata,m.,nagase,h.,endo,t.,ueda,h.,glimepiride.actacrystallogr;1997,c53(3):329–331.):采用氯仿溶解粗品,再加入乙醇析晶得到。由于精制使用到氯仿,其限度为60ppm,限度比较小,实验证明,很难将氯仿控制在限度范围内。因此,该方法不适合作为格列美脲原料药的生产工艺。
最早公开的晶型ⅱ制备方法(endot,iwatam,nagaseh,shirom,uedah.stppharmasci;2003,13(4):281–286.):采用乙醇和水升温溶解粗品,再经过冷却后析晶得到。由于格列美脲即使在热的乙醇和水体系中是微溶,按文献方法所用的溶剂量大,析晶时间长,效率低。因此不适合于大生产制备晶型ⅱ。此外文献提及晶型ⅱ向晶型i转换,只需要将晶型ⅱ加热到140℃就能转换成晶型i。
文献internationaljournalofpharmaceutics,553:272–280,2018;journalofpharmaceuticalsciences,101(2):794-804,2012报道了晶型ⅱ的制备方法,与上述制备方法基本一致,2g样品需要1000ml乙醇加热溶解,再加入500ml水,再需要降温至25保温12小时,再降至5℃保温7天,才能得到晶型ⅱ产品。这种方法使用溶剂量大,耗时长,效率低。无法应用到大生产。
晶型i是适合药用的晶型,目前没有文献记载其能应用到大生产的制备方法,以及如何解决溶残合格的问题。
实验发现,晶型i比较容易得到,但在析晶过程中非常容易造成溶剂包裹,这种包裹的溶剂十分稳定,采用烘料、增加烘料时间、真空干燥、气流粉碎后再烘等方式,都无法将其去除或降低到合格限度内(ichq3c限度标准)。
经过实验,晶型ⅱ在140℃以上高温条件能转化为晶型i,但由于转晶温度太高,格列美脲容易降解变质和变色,其主要降解杂质为如下式(式2化合物),并超标(大于0.4%)。因此,这个转晶工艺在格列美脲原料药的生产方面,没有实用价值。
技术实现要素:
针对现有工艺的缺陷,提供一种质量好,能够满足药典要求的格列美脲晶型i。为制剂提供优良的原料药。
本发明的技术方案是:
一种格列美脲晶型i的制备方法,包括以下步骤:
第一步将格列美脲粗品溶解于醇的水溶液中,加适量氨水,搅拌溶解,所述醇选自乙醇、异丙醇或正丙醇中的一种。
所述醇不包括甲醇、叔丁醇。因甲醇和叔丁醇所得的晶型为晶型i,而不是晶型ⅱ。
优选的,所述适量的氨水,指氨水与格列美脲的摩尔比在2.0~2.5:1范围。
第二步过滤,向滤液中加入乙酸,中和滤液中的氨,得格列美脲晶型ⅱ。
本步骤中乙酸与氨水的摩尔比不低于1:1,以保证氨能完全被中和。
第三步向晶型ⅱ中,加入质量为其4~8倍的酮类溶剂,在10~35℃温度范围内,搅拌10h以上,过滤,70℃烘干,得格列美脲晶型i产品。
优选的所述酮类溶剂选自丙酮或丁酮中的一种。
本发明技术方案中,晶型ⅱ颗粒大小不影响转晶效果,但影响转晶的速度。颗粒越大,转晶所需要的时间越长。所制备的晶型ⅱ可以经过粉碎得到更小颗粒,可明显提高转晶的速度。
附图说明
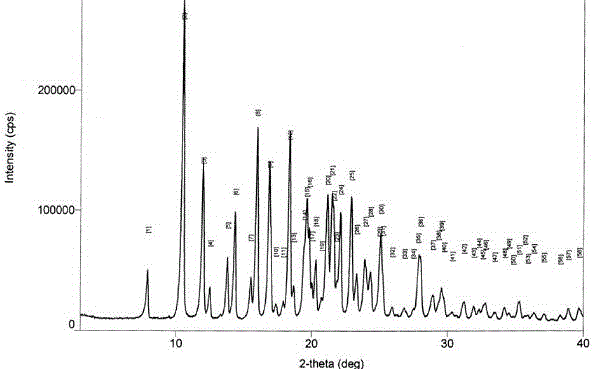
图1晶型ⅱ的xrd图谱。
图2晶型i的xrd图谱。
图3晶型ⅱ的dsc图谱。
图4晶型i的dsc图谱。
有益效果:通过将格列美脲粗品制备为晶型ⅱ,再转换为晶型i的方式,本发明获得了高质量的格列美脲晶型i原料药,其溶剂残留符合ichq3c限度标准,杂质化合物2未见明显的增长。
实施例和对照例
本发明实施例和对照例用粗品,hplc纯度为99.56%,丙酮溶残1.63%。
一步转晶法实施
对照例1(丙酮)
将所制备得到的列美脲粗品5.0g,加入丙酮50.0g,纯化水25.0g,27%氨水1.61g,溶清过滤,向滤液中加滴1.68g冰乙酸(用10.0g丙酮稀释),滴毕,控温25℃搅拌1小时,过滤,70℃干燥至恒重,得格列美脲晶型i4.26g,收率85.2%;hplc纯度为99.81%;溶剂残留为丙酮,含量1.27%(丙酮限度为0.5%),超过限度。
对照例2(甲醇)
将所制备得到的列美脲粗品10.0g,加入甲醇60.0g,27%氨水3.21g,纯化水20.0g溶清过滤,向滤液中加滴3.37g冰乙酸(用20.0g甲醇稀释),滴毕,控温25℃搅拌2小时,过滤,70℃干燥至恒重,得到格列美脲i晶型9.25g,收率92.5%;hplc纯度为99.76%;溶剂残留为甲醇,含量0.62%(甲醇限度为0.3%),超过限度。
两步转晶法实施例
晶型ⅱ的制备
实施例1(乙醇)
将格列美脲粗品13.0g,加入95%乙醇130g,27%氨水4.18g,水25g,搅拌溶解,溶清,过滤,向滤液慢慢加入4.38g冰乙酸(用26g水稀释),滴加完毕,继续搅拌1小时,过滤得,水洗,70℃干燥,得产品12.27g,收率:94.4%。经xrd检测,判定为晶型ⅱ。
实施例2(正丙醇)
将格列美脲粗品10.0g,加入正丙醇100g,27%氨水3.21g,水20g,搅拌溶解,溶清,过滤,向滤液慢慢加入3.37g冰乙酸(用20g水稀释),滴加完毕,继续搅拌1小时,过滤,水洗,70℃干燥,得产品8.47g,收率:84.7%。经xrd检测,判定为晶型ⅱ。
实施例3(异丙醇)
将格列美脲粗品5.0g,加入异丙醇30g,纯化水10g,27%氨水1.61g,搅拌溶解,溶清,过滤,向滤液慢慢加入1.68冰乙酸(用10g水稀释),滴加完毕,继续搅拌1小时,过滤,水洗,70℃干燥,得产品4.65g,收率:93.0%。经xrd检测,判定为晶型ⅱ。
转晶制备晶型i
实施例4
将所制备得到的列美脲ⅱ晶型5.0g,加入丙酮25.0g,控温35℃搅拌20小时,过滤,水洗,80℃干燥至恒重,得到格列美脲4.56g,收率91.2%;hplc纯度为99.92%;溶剂残留只有丙酮,检测值为0.21%(限度为0.5%)。经xrd检测,显示为ⅰ晶型。
实施例5
将所制备得到的列美脲ⅱ晶型,经万能粉碎机粉碎,取4.0g,加入丙酮20.0g,控温25℃搅拌10小时,过滤,水洗,80℃干燥至恒重,得到格列美脲4.59g,收率91.8%,hplc纯度为99.96%,溶剂残留只有丙酮0.09%(限度为0.5%)。经xrd检测,显示为ⅰ晶型。
实施例6
将所制备得到的列美脲ⅱ晶型5.0g,加入丁酮25.0g,控温10℃搅拌30小时,过滤,水洗,80℃干燥至恒重,得到格列美脲4.66g,收率93.1%,hplc纯度为99.90%,溶剂残留只有丁酮0.13%(限度为0.5%)。经xrd检测,显示为ⅰ晶型。
- 还没有人留言评论。精彩留言会获得点赞!