一类具有联苯酚基取代的苯并三氮唑类化合物及其制备与应用的制作方法
本发明涉及有机材料、光吸收剂领域,具体涉及一类新型的紫外-短波长蓝光吸收剂,[3-(2h-苯并[d][1,2,3]三氮唑-2-基)-4',5-二取代-[1,1'-联苯]-2-酚]的制备方法和作为有机光吸收材料的应用。
背景技术:
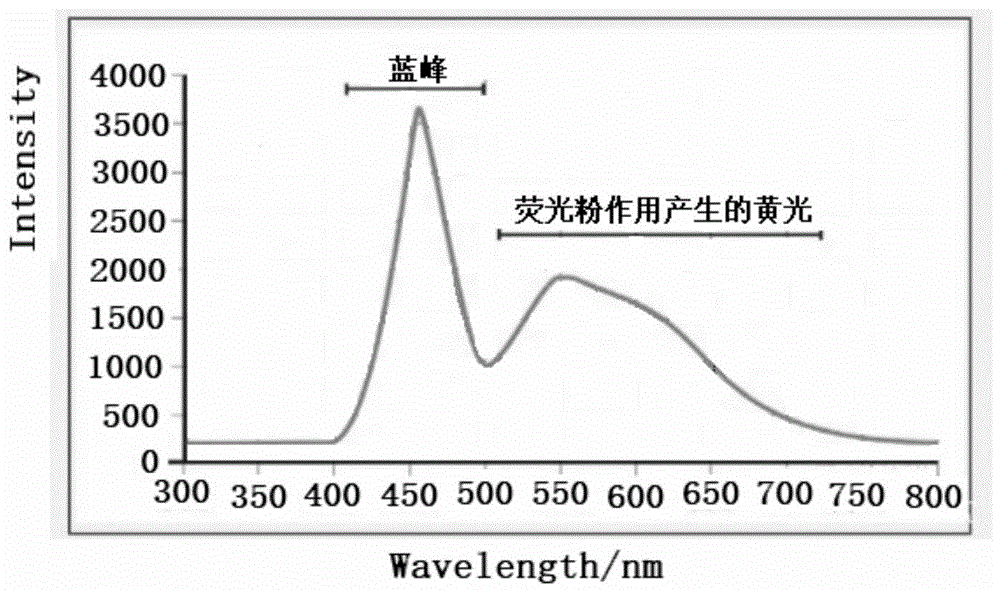
:人类肉眼可见的光的波长约为400~700nm。400~500nm为蓝光,在可见光中波长最短,能量最高。人的眼睛中色素最多的部位是黄斑,它决定了一个人的视力好坏。研究结果表明,人眼最不能接受的是蓝光和紫外光。蓝光杀伤人眼活性细胞的能力是绿光的10倍。蓝光能够直达视网膜,如果长期接触大量低波长的蓝光,就会造成大量人眼活性细胞被杀死,从而对视力造成不可逆的损伤。特别需要强调的是,由于儿童的视网膜黄斑对蓝光的吸收能力弱,同样的光照强度情况下,透过的蓝光辐射量是成年人的两倍左右,所以蓝光辐射对儿童视网膜的危害更大。处于生长发育期的低龄人群若长期使用这些电子产品,轻者会诱发致盲眼病,重者会造成终身的视力低下,甚至全盲。目前,家庭和办公室所使用的照明灯、手机、电脑、平板电脑、电视等几乎都是采用的led光源。该光源通常是由主峰为465nm的蓝光和黄光组成。led光源所发出的光的光谱曲线如图1所示。从图1看出,我们通常接触到的电子产品所发出的光,波长在450nm的光占有很大比例。所以我们每天都在接受着大量波长在450nm的光的辐射。为了减轻或阻止蓝光对眼睛的影响,有必要研发一类材料,应用于眼镜、手机、电脑屏幕、电视机等产品,吸收高能量的紫外光和部分短波长蓝光,即把有害的短波长的光消除掉,从而起到保护我们的眼睛的目的。技术实现要素:本发明的目的是提供一类带有苯并三氮唑类结构单元的新型有机吸光剂材料,并进一步地研究了这类材料合成路径及实际应用。本发明是通过以下技术方案予以实施:本发明首先提供了一类具有联苯酚基取代的苯并三氮唑类化合物,其化学结构通式为:所述r1为选自烷基、烷氧基、芳基、氨基或卤素;当所述r3为氢时,所述r2为选自烷基、烷氧基、芳基、氨基或卤素,化学名为[3-(2h-苯并[d][1,2,3]三氮唑-2-基)-4',5-二取代-[1,1'-联苯]-2-酚],结构式如式ⅰ:当所述r3为甲氧基时,所述r2为苯或4-取代苯基,结构式如式ⅱ:优选地,所述r1和r2所选的基团中,烷基是指c1~c7的烷基,芳基是指含有1~4个芳环的基团。本发明提供的具有联苯酚基取代的苯并三氮唑类化合物,具有紫外吸收剂的结构单元,即具有可以形成稳定分子内氢键的结构单元,凭借这种氢键的形成和断裂,可以消除紫外线的破坏作用,因此对紫光可表现出特异的稳定性;同时其光吸收主峰比常见的苯并三氮唑紫外吸收剂(uv-p和uv-329)红移约10nm,主吸收峰处在360nm左右,可满足某些特定行业的使用要求。这类吸收剂的吸收波长的覆盖范围达到400nm;提高其使用量时,其最大吸收波长的覆盖范围会超过420nm;具有吸收短波长蓝光的特性,用作紫外-短波长蓝光吸收剂并使于多个方面,如:具体地可制成涂料或制成薄膜,涂覆或贴覆于能所产生高能量的紫外光和部分短波长蓝光的产品的可视界面上、或眼镜镜片上。本发明提供的具有联苯酚基取代的苯并三氮唑类化合物,其制备方法如下:采用2-(2ˊ-羟基-4ˊ-取代-5ˊ-烷基苯基)苯并三氮唑结构类物质作为原料,其中所述的4ˊ-取代位置基团为选自氢、烷基、烷氧基或芳基;优选采用uv-p或uv-329作为原料;采用有机溶剂将原料溶解后,加入溴素,充分混合发生溴代反应,得到溴代产物2-(2ˊ-羟基-3ˊ-溴-4ˊ-取代-5ˊ-烷基苯基)苯并三氮唑;再将溴代产物2-(2ˊ-羟基-3ˊ-溴-4ˊ-取代-5ˊ-烷基苯基)苯并三氮唑与芳基硼酸或芳基硼酸酯混合溶解,在催化剂和碱性条件下,进行偶合反应,即得最终产物。所述uv-p为2-(2ˊ-羟基-5ˊ-甲基苯基)苯并三氮唑,其结构式为所述uv-329为2-(2ˊ-羟基-5ˊ-异辛基苯基)苯并三氮唑,其结构式为具体合成路线如下:以2-(2ˊ-羟基-4ˊ-取代-5ˊ-烷基苯基)苯并三氮唑结构类物质为原料,优选以uv-p或uv-329为原料,依次经过第一步反应溴代和第二步偶合反应完成,得到目标化合物:优选地,所述溴代反应中,溶解原料的有机溶剂为选自二氯甲烷、三氯甲烷、四氯化碳或乙酸中的一种;原料分别与溴素的摩尔比为1:(0.9~1.8);反应温度为-40℃~室温(约25℃),反应时间为5~12小时。优选地,所述偶合反应中,所述溴代产物2-(2ˊ-羟基-3ˊ-溴-4ˊ-取代-5ˊ-烷基苯基)苯并三氮唑与芳基硼酸或芳基硼酸酯的摩尔比为1:(0.9~1.6)。进一步地,以下列举出本发明优选涉及的化合物,其结构如表1所示,表1所列举的化合物将在实施例中进一步进行表征。须说明的是,本发明的保护范围不仅限于表1所列举的化合物,只要符合本发明提出的结构通式的化合物均在本发明的保护范围内。表1本专利涉及的化合物的结构本专利所涉及的化合物,大部分光吸收主峰处在350nm附近,比常见的苯并三氮唑紫外吸收剂(uv-p和uv-329)的主吸收峰339.5nm红移约10nm,吸收波长明显增加,可满足某些特定行业的使用要求。本发明所涉及的某些吸收剂吸收波长的覆盖范围达到400nm;提高其使用量时,其吸收波长的覆盖范围会超过420nm;具有吸收短波长蓝光的特性,可作为蓝光吸收剂使用。附图说明图1为市场上led光源的光谱曲线图;图2为化合物uv-329在二氯甲烷中的紫外吸收图;图3为化合物uv-p在二氯甲烷中的紫外吸收图;图4为实施例4的产物在二氯甲烷中的紫外吸收图;图5为实施例6的产物在二氯甲烷中的紫外吸收图;图6为实施例9的产物在二氯甲烷中的紫外吸收图;图7为实施例12的产物在二氯甲烷中的紫外吸收图;图8为实施例13的产物在二氯甲烷中的紫外吸收图。具体实施方式实施例1本实施例旨在给出溴代反应的合成方法以及溴代反应的产物。化合物:2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑,合成方法:在100ml的圆底烧瓶中,分别加入紫外吸收剂uv-p(2-(2'-羟基-5'-甲基苯基)苯并三氮唑4.50g(20mmol)、二氯乙烷40ml,搅拌使其溶解;慢慢滴加溴素3.84g(24mmol),室温搅拌下反应8小时;把混合物导入水中,慢慢用固体碳酸氢钠中和至中性;分离出有机层,水层用二氯乙烷(10ml)重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到目标产物5.59克,产率92%。产物熔点为202.4~202.9℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.88(s,1h),8.19(d,j=1.3hz,1h),7.93(dd,j=6.6,3.1hz,2h),7.50(dd,j=6.6,3.1hz,2h),7.45(d,j=1.5hz,1h),2.39(s,3h).13cnmr(100mhz,cdcl3)δ:144.8,142.8,134.5,130.4,128.1,125.4,120.6,117.7,112.1,20.4.esi-ms,m/z304.0[m+h]+。实施例2本实施例是以实施例1的产物为反应原料,旨在给出偶合反应的合成方法以及偶合反应的产物。化合物:2-(2'-羟基-3'-苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯40ml、乙醇8ml和水16ml混合液的100ml三颈瓶中依次加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、苯硼酸1.22g(10mmol)和碳酸钾5.52g(40mmol),磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到目标产物2.71克,产率90%,产物熔点202.4~202.9℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.58(s,1h),8.22(d,j=2.0hz,1h),7.91(dd,j=6.5,3.1hz,2h),7.65(d,j=7.5hz,2h),7.51–7.41(m,4h),7.37(t,j=7.4hz,1h),7.23(d,j=2.1hz,1h),2.43(s,3h).13cnmr(100mhz,cdcl3)δ:145.0,142.8,137.8,132.6,131.7,129.5,129.2,128.1,127.7,127.4,125.3,120.6,117.7,20.6.esi-ms,m/z302.1[m+h]+,324.1[m+na]+。实施例3本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-二苯氨基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-硼酸三苯胺2.89g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物4.15克,产物产率89%,产物熔点213.5~213.8℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.69(s,1h),8.21(d,j=1.5hz,1h),7.93(dd,j=6.6,3.1hz,2h),7.56(d,j=8.6hz,2h),7.48(dd,j=6.6,3.1hz,2h),7.29(s,1h),7.27(s,2h),7.16(dd,j=8.1,4.3hz,7h),7.03(t,j=7.3hz,3h),2.44(s,3h).13cnmr(100mhz,cdcl3)δ:147.7,147.0,145.0,142.8,132.3,131.7,131.2,130.2,129.2,127.7,124.5,123.2,122.9,120.2,117.6,20.7.esi-ms,m/z507.2[m+na]+.实施例4本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-苯基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-联苯硼酸1.98g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物3.27克,产率87%,产物熔点217.8~218.3℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.68(s,1h),8.26(d,j=1.5hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.75(d,j=8.5hz,2h),7.72–7.67(m,3h),7.66(s,1h),7.51–7.44(m,4h),7.37(t,j=7.4hz,1h),7.29(d,j=2.1hz,1h),2.46(s,3h).13cnmr(100mhz,cdcl3)δ:145.1,142.8,141.0,140.2,136.7,134.2,134.1,132.4,131.20,129.9,129.5,129.3,128.8,127.8,127.3,127.2,126.9,125.4,120.7,118.3,117.7,20.7.esi-ms,m/z376.3[m-h]-.实施例5本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-氰基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-氰基苯硼酸1.47g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物2.93克,产率90%,产物熔点:217.8~218.3℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.76(s,1h),8.31(d,j=1.5hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.79(d,j=8.6hz,2h),7.75(d,j=8.5hz,2h),7.51(dd,j=6.6,3.1hz,2h),7.22(d,j=2.1hz,1h),2.46(s,3h).13cnmr(100mhz,cdcl3)δ:145.0,142.8,142.6,132.1,131.9,130.2,129.6,129.5,128.0,125.5,121.7,119.1,117.7,111.0,20.6.esi-ms,m/z325.1[m-h]-.实施例6本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-乙酰基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-乙酰基苯硼酸1.64g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物2.90克,产率85%,产物熔点195.9~197.8℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.71(s,1h),8.28(d,j=2.0hz,1h),8.06(d,j=8.5hz,2h),7.94(dd,j=6.6,3.1hz,2h),7.77(d,j=8.5hz,2h),7.50(dd,j=6.6,3.1hz,2h),7.25(d,j=2.0hz,1h),2.66(s,3h),2.46(s,3h).13cnmr(100mhz,cdcl3)δ:197.9,145.1,142.8,142.7,135.9,132.3,130.4,129.7,129.5,128.2,127.9,125.4,121.4,117.7,26.7,20.6.esi-ms,m/z344.1[m+h]+.实施例7本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-甲氧基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-甲氧基苯硼酸1.52g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物2.70克,产率82%,产物熔点157.9~159.6℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.58(s,1h),8.20–8.16(m,1h),7.90(dd,j=6.5,3.1hz,2h),7.60(d,j=8.7hz,2h),7.45(dd,j=6.6,3.0hz,2h),7.20(d,j=2.0hz,1h),7.00(d,j=8.8hz,2h),3.86(s,3h),2.42(s,3h).13cnmr(100mhz,cdcl3)δ:158.98,144.97,142.73,132.28,131.25,130.59,130.12,129.14,127.64,125.25,120.13,117.60,113.60,55.30,20.62.esi-ms,m/z332.1[m+h]+.实施例8本实施例是以其他常用的紫外吸收剂作为溴代反应的原料,溴代反应的方法与实施例1相同;旨在给出以其他紫外吸收剂作为原料的溴代反应产物。化合物:2-(2'-羟基-3'-(4-甲氧基-3-溴)苯基-5'-甲基苯基)苯并三氮唑,合成方法:在100ml的圆底烧瓶中,分别加入紫外吸收剂2-(2'-羟基-3'-(4-甲氧基)苯基-5'-甲基苯基)苯并三氮唑6.62g(20mmol)、二氯乙烷40ml,搅拌使其溶解;慢慢滴加溴素3.84g(24mmol),室温搅拌下反应8小时;把混合物导入水中,慢慢用固体碳酸氢钠中和至中性;分离出有机层,水层用二氯乙烷(10ml)重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到目标产物8.05克,产率为98%,产物熔点156.3~157.1℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.65(s,1h),8.23(d,j=1.5hz,1h),7.94(dd,j=6.6,3.0hz,2h),7.88(d,j=2.2hz,1h),7.60(dd,j=8.5,2.2hz,1h),7.50(dd,j=6.6,3.1hz,2h),7.20(d,j=1.8hz,1h),7.00(d,j=8.5hz,1h),3.96(s,3h),2.44(s,3h).13cnmr(100mhz,cdcl3)δ:155.15,144.93,142.76,134.24,132.12,131.46,129.78,129.59,129.30,127.78,125.31,120.61,117.63,111.47,111.29,76.71,56.33,20.62.esi-ms,m/z410.0[m+h]+.实施例9本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-叔丁基)苯基-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、4-叔丁基苯硼酸1.78g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物3.16克,产率89%,产物熔点174.4~175.5℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.63(s,1h),8.21(d,j=1.5hz,1h),7.91(dd,j=6.6,3.1hz,2h),7.61(d,j=7.5hz,2h),7.53–7.42(m,5h),2.43(s,3h),1.40–1.37(m,9h).13cnmr(100mhz,cdcl3)δ:150.22,145.03,142.74,134.75,132.50,131.53,129.17,129.08,127.66,125.26,125.09,120.33,117.61,34.59,31.39,20.62.esi-ms,m/z358.2[m+h]+.实施例10本实施例采用实施例1的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(2-萘基)-5'-甲基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-甲基苯基)苯并三氮唑3.03g(10mmol)、2-萘硼酸1.72g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物3.15克,产率90%,产物熔点165.1~166.0℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.68(s,1h),8.26(d,j=1.5hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.75(d,j=8.5hz,2h),7.72–7.67(m,3h),7.66(s,1h),7.51–7.44(m,4h),7.37(t,j=7.4hz,1h),7.29(d,j=2.1hz,1h),2.46(s,3h).13cnmr(100mhz,cdcl3)δ:145.21,142.76,135.37,133.41,132.73,132.65,131.55,129.32,128.26,128.18,127.80,127.72,127.62,127.39,125.99,125.96,125.33,120.67,117.62,20.65.esi-ms,m/z352.1[m+h]+.实施例11本实施例是以uv-329取代uv-p作为溴代反应的原料,溴代反应的方法与实施例1相同;旨在给出以uv-329作为原料的溴代反应产物。化合物:2-(2'-羟基-3'-溴-5'-异辛基苯基)苯并三氮唑,合成方法:在100ml的圆底烧瓶中,分别加入紫外吸收剂2-(2'-羟基-5'-异辛基苯基)苯并三氮唑)(即uv-329)6.46g(20mmol)、二氯乙烷40ml,搅拌使其溶解;慢慢滴加溴素3.84g(24mmol),室温搅拌下反应8小时;把混合物导入水中,慢慢用固体碳酸氢钠中和至中性;分离出有机层,水层用二氯乙烷(10ml)重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到得到目标产物7.65克,产率为95%,产物熔点131.9~133.3℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.95(s,1h),8.40(d,j=2.3hz,1h),7.95(dd,j=6.6,3.1hz,2h),7.63(d,j=2.3hz,1h),7.50(dd,j=6.6,3.1hz,2h),1.79(s,2h),1.44(s,6h),0.79(s,9h).13cnmr(100mhz,cdcl3)δ:144.56,143.18,142.81,131.98,127.98,125.22,118.07,117.71,111.91,56.74,38.41,32.45,31.91,31.52.esi-ms,m/z402.1[m+h]+.实施例12本实施例以实施例11的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-甲氧基苯基)-溴-5'-异辛基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-异辛基苯基)苯并三氮唑4.01g(10mmol)、4-甲氧基苯硼酸1.52g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物2.84克,产率81%,产物熔点156.4~159.0℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.67(s,1h),8.40(d,j=2.4hz,1h),7.92(dd,j=6.6,3.1hz,2h),7.61(d,j=8.7hz,2h),7.48–7.41(m,3h),7.03(d,j=8.8hz,2h),3.87(s,3h),1.83(s,2h),1.48(s,6h),0.81(s,9h).13cnmr(100mhz,cdcl3)δ:158.96,144.83,142.76,141.74,130.67,130.64,129.86,127.58,125.01,117.66,113.64,56.85,55.36,38.34,32.48,31.95,31.67.esi-ms,m/z430.2[m+h]+.实施例13本实施例以实施例11的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-二苯氨基苯基)-溴-5'-异辛基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-异辛基苯基)苯并三氮唑4.01g(10mmol)、4-硼酸三苯胺2.89g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物4.50克,产率80%,产物熔点179.6~179.7℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.76(s,1h),8.40(d,j=2.4hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.56(d,j=8.6hz,2h),7.49(dd,j=6.6,3.1hz,2h),7.45(d,j=2.4hz,1h),7.29(d,j=8.4hz,4h),7.18(dd,j=8.0,4.1hz,6h),7.04(t,j=7.3hz,2h),1.83(s,2h),1.49(s,6h),0.81(s,9h).13cnmr(100mhz,cdcl3)δ:144.83,142.74,141.78,130.50,130.31,129.78,129.24,127.60,125.03,124.55,123.14,122.91,117.74,117.64,56.86,38.35,32.48,31.95,31.67.esi-ms,m/z567.3[m+h]+.实施例14本实施例以实施例11的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-氰基苯基)-5'-异辛基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-溴-5'-异辛基苯基)苯并三氮唑4.01g(10mmol)、4-氰基苯硼酸1.47g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物3.38克,产率80%,产物熔点177.0~178.1℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.82(s,1h),8.50(d,j=2.4hz,1h),7.96(dd,j=6.6,3.0hz,2h),7.77(d,j=3.1hz,4h),7.51(dd,j=6.6,3.1hz,2h),7.40(d,j=2.4hz,1h),1.84(s,2h),1.49(s,6h),0.81(s,9h).13cnmr(100mhz,cdcl3)δ:144.81,143.03,142.80,142.28,131.92,130.33,129.53,128.98,127.90,125.24,119.26,119.09,117.69,110.90,56.85,38.41,32.50,31.96,31.63.esi-ms,m/z425.2[m+h]+.实施例15本实施例是以其他常用的紫外吸收剂作为溴代反应的原料,溴代反应的方法与实施例1相同;旨在给出以其他紫外吸收剂作为原料的溴代反应产物。本实施例以实施例11的溴代产物作为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-甲氧基-3-溴苯基)-5'-异辛基苯基)苯并三氮唑,合成方法:在100ml的圆底烧瓶中,分别加入紫外吸收剂2-(2'-羟基-3'-(4-甲氧基苯基-5'-(异辛基)苯基)苯并三氮唑8.58g(20mmol)、二氯乙烷40ml,搅拌使其溶解;慢慢滴加溴素3.84g(24mmol),室温搅拌下反应8小时;把混合物导入水中,慢慢用固体碳酸氢钠中和至中性;分离出有机层,水层用二氯乙烷(10ml)重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到目标产物7.90克,产率为78%,产物熔点145.3~146.1℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.70(s,1h),8.42(d,j=2.4hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.87(d,j=2.1hz,1h),7.58(dd,j=8.5,2.1hz,1h),7.49(dd,j=6.6,3.1hz,2h),7.38(d,j=2.4hz,1h),7.02(d,j=8.5hz,1h),3.96(s,3h),1.83(s,2h),1.48(s,6h),0.81(s,9h).13cnmr(100mhz,cdcl3)δ:155.16,144.78,142.76,141.93,134.31,131.99,129.66,129.60,129.24,127.69,125.06,118.16,117.66,111.53,111.33,56.84,56.36,38.36,32.48,31.96,31.66.esi-ms,m/z508.3[m+h]+.实施例16本实施例是实施例15所得的合成产物为原料,进行偶合反应,偶合反应条件与实施例2相同。化合物:2-(2'-羟基-3'-(4-甲氧基-3-(4-甲氧基苯基))-5'-异辛基苯基)苯并三氮唑,合成方法:往盛有甲苯、乙醇和水混合液的三颈瓶中加入2-(2'-羟基-3'-(4-甲氧基-3-溴苯基)-5'-(异辛基苯基)苯并三氮唑5.07g(10mmol)、4-甲氧基苯硼酸1.52g(10mmol)、以及碳酸钾40mmol,磁力搅拌溶解;再将氮气在溶液中剧烈鼓泡5min,加入四(三苯基膦)钯0.3g(0.26mmol)继续用氮气在溶液中剧烈鼓泡5min;在氮气环境下反应回流24小时,待反应完全后,将反应混合液冷却至室温,分离出有机层,水层用二氯乙烷10ml重复萃取2次;合并有机层,用无水硫酸钠干燥,旋转蒸发除去溶剂后,用二氯乙烷重结晶得到偶合得到目标产物3.85克,产率72%,产物熔点75.5~76.1℃。化合物核磁共振表征:1hnmr(400mhz,cdcl3)δ:11.67(s,1h),8.41(d,j=2.3hz,1h),7.94(dd,j=6.6,3.1hz,2h),7.64–7.55(m,4h),7.48(dd,j=6.6,3.1hz,2h),7.45(d,j=2.2hz,1h),7.09(d,j=8.2hz,1h),6.98(d,j=8.7hz,2h),3.87(d,j=13.0hz,6h),1.82(s,2h),1.48(s,6h),0.81(s,9h).13cnmr(100mhz,cdcl3)δ:158.71,155.88,144.87,142.76,141.82,131.95,130.90,130.74,130.59,130.01,129.81,129.27,127.59,125.03,117.75,117.65,113.54,110.96,56.85,55.73,55.28,38.36,32.48,31.95,31.66.esi-ms,m/z536.3[m+h]+.试验例17将实施例1~16依次作为样品1~16、uv-p作为样品17、以及uv-329作为样品18,进行检测试验。检测方法如下:取样品一定量的样品(毫克量)于10ml容量瓶中,用二氯甲烷定溶至10ml;取该溶液0.1ml再定溶至10ml。然后用2ml的比色皿测定紫外光谱和相应的最大吸收波长处的吸光度,获得样品的最大吸收波长;通过样品的摩尔浓度数据,计算获得最大吸收波长处的摩尔吸光度(即摩尔吸光系数)。同时,为了对比,也在同样条件下测定了紫外吸收剂uv-p和uv-329的相应数据。uv-p、uv-329和样品4、6、9、12和13在二氯甲烷中的紫外吸收图见附图2~8。本专利所涉及的化合物在二氯甲烷中的最大吸收波长和对于的即摩尔吸光系数(ξ)列于表2。表2本专利所涉及的化合物在二氯甲烷中的最大吸收波长和对于的即摩尔吸光系数样品分子量最大吸收波长/nm吸光度摩尔吸收度(cm2·mol-1)x10-51303.00338.00.3170.3098422301.12346.00.2610.2535243468.20307.00.4430.4413034377.15346.00.2470.2328905326.12347.00.2610.2364376343.13348.00.2880.2823477331.13349.00.2290.2297848409.04345.00.2060.2055189357.18346.50.2070.20537910351.14346.00.2150.20970911401.11339.00.1620.16661512429.24348.00.1980.20235613566.30308.00.4780.48337814424.23348.00.2610.25749815507.15341.00.2230.22618916535.28347.00.2210.22749417225.1339.50.3800.32897818323.2339.50.2810.283810对比uv-p、uv-329和化合物4、6、9、12和13在二氯甲烷中的紫外吸收谱图和表2中所列出的化合物的紫外吸收数据发现,本专利所涉及的光吸收剂,某些光吸收主峰处在350nm附近,比常见的苯并三氮唑紫外吸收剂(uv-p和uv-329)的主吸收峰339.5nm红移约10nm,吸收波长明显增加,可满足某些特定行业的使用要求。本发明所涉及的某些吸收剂吸收波长的覆盖范围达到400nm;提高其使用量时,其吸收波长的覆盖范围会超过420nm;具有吸收短波长蓝光的特性,可作为蓝光吸收剂使用。当前第1页12
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1