一种氧化氮硫环戊烷化合物的合成方法与流程
[0001]
本发明涉及一种氧化氮硫环戊烷化合物的合成方法。
背景技术:
[0002]
2-氧化四氢噻唑1,3(化合物1),它是一种在医药、农业、轻化工等领域中使用的重要化学品中间体。其制备方法有多种。但在这些方法中,有的对环境不友好,存在安全生产问题;有的反应条件过分苛刻、或原料很难获得、或产物收率低、纯度难以满足要求,不易得到廉价、优质的大量产品。因此,这些方法在实际工业化生产中都存在困难。
[0003]
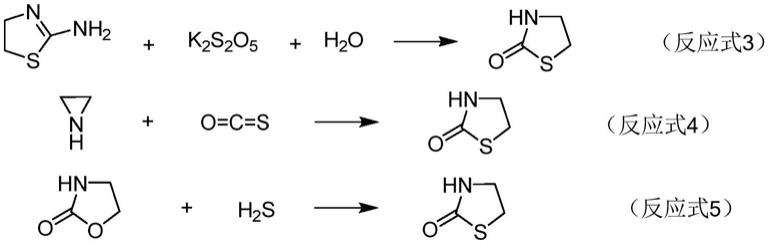
如,申请人的一项授权专利(专利号:cn1403447)用化合物2和环氧乙烷在水中进行反应,得到化合物1(反应式1)。该制备方法虽已用于工业化生产,但由于副产物环硫乙烷在反应条件下会发生聚合反应生成聚环硫乙烷或环硫环氧乙烷的共聚物,因此给环境造成大量固体废物,而且环氧乙烷的安全生产问题也使工业生产造成困难。
[0004][0005]
又如,申请人曾试用文献(沈德隆现代农药2003,2(3):11)的方法(反应式2)进行实验室研究验证,实验过程中发现,产品的纯度难以达到gc96%以上的纯度要求。
[0006][0007]
在文献中也有使用化合物2做起始原料制备化合物1的类似方法,如将化合物2与氯乙醇反应,其反应转化率低,收率不足50%,副反应多,后处理比较困难,效果不够理想。
[0008]
除此之外,在文献或者从理论上设计还有几条合成路线,如:
[0009][0010][0011]
在上述几条路线中,k2s2o5是少见化合物,cso和h2s均是气体生产规模难以实施的气体,文献中没有详细研究报道表明它们已用于生产信息。看起来只有(反应式6)可能用于生产,但由于原料cocl2是有毒气体,因此,生产使用可能性很小。
[0012]
总结以上背景,现有技术中虽然制备化合物1的方法很多,但是能够用于实际工业
化生产的却很少。
[0013]
目前化合物1的合成和生产主要利用半胱胺盐酸盐为原料与碳酸双(三氯甲)酯反应(反应式7),或用半胱胺盐酸盐与尿素反应(反应式8)的合成方法。
[0014]
由于半胱胺盐酸盐纯度好,而碳酸双(三氯甲)酯是强有力的酰化剂,性质稳定,极少的挥发性,反应条件温和,反应后产生的hcl可在反应中加入碱的方法中和而不会释放在大气中,因而广为生产者欢迎。其操作方法一般是将半胱胺盐酸盐溶于水中,将碳酸双(三氯甲)酯溶解在有机溶剂中,在碱性条件下(如氢氧化钾或氢氧化钠中)滴入上述有机溶剂,在低温(如10~20℃)下进行反应,反应完毕后用有机溶剂提取纯化。另一种合成方法是将半胱胺盐酸盐和尿素混合在180℃反应至氨气放尽。两种方法反应后的后处理是一样的。
[0015][0016]
虽然利用半胱胺盐酸盐为原料与碳酸双(三氯甲)酯反应,或用半胱胺盐酸盐与尿素反应制备化合物1的方法已经比较成熟。但是,该反应的成本十分昂贵。因为半胱胺盐酸盐的价格十分昂贵,它几乎占了原料费用的75%,因为,目前市售半胱胺盐酸盐几乎都用(反应式12)的方法生产,虽然这一反应制备得到的半胱胺盐酸盐质量好、收率高,但这一反应路线中既要用易燃易爆的二硫化碳,还要在盐酸沸点条件下进行240小时左右的水解反应,花费大量人力、物力、时间、环境污染也很大。由此可见获得廉价优质的半胱胺的研究显得非常重要。申请人为此做了大量工作研究。
[0017][0018]
首先,申请人选用了理论上最简单的方法作为研究,用乙醇胺与na2s直接反应,如在氢氧化钠或乙醇钠的条件下进行亲核取代反应(反应式13),希望获得廉价半胱胺。
[0019]
h2nch2ch2oh+na2s
→
h2nch2ch2sh
ꢀꢀ
(反应式13)
[0020]
但在实际操作中,这一反应虽然能够进行,但转化率较低,最重要的问题是反应生产的半胱胺及未反应的原料在碱性条件下发生聚合或进一步的亲核反应,导致无法从反应液中分离出理想量的半胱胺。
[0021]
为了增加反应活性,申请人将乙醇胺中的羟基进行酯化,或将乙醇胺中的羟基用氯、溴等卤素取代(如h2nch2ch2oso3、h2nch2ch2oso2c6h4ch3、h2nch2ch2cl),但是还是很难分离出纯度高、收率好的产品。申请人也尝试过其他许多合成路线,做了大量研究,均未能得到廉价的半胱胺,找到一种低成本生产化合物2-氧化四氢噻唑1,3的方法。
技术实现要素:
[0022]
为了解决上述问题,找到一种低成本生产化合物2-氧化四氢噻唑1,3的方法,本发明提供了一种氧化氮硫环戊烷化合物的合成方法,它是由乙醇胺硫酸酯、硫化碱、无机碱和碳酸双(三氯甲)酯经过一步法反应即得;
[0023]
其中,所述的氧化氮硫环戊烷化合物为式i所示:
[0024][0025]
进一步地,所述的一步法反应为将乙醇胺硫酸酯、硫化碱和无机碱加入溶剂后,在反应温度下滴加含碳酸双(三氯甲)酯的溶液进行反应。
[0026]
进一步地,所述的乙醇胺硫酸酯、硫化碱、无机碱和碳酸双(三氯甲)酯的质量比为28:10~30:1~20:10~20;所述的溶剂与乙醇胺硫酸酯的体积质量比为28:60~100(v/w);所述的含碳酸双(三氯甲)酯的溶液中碳酸双(三氯甲)酯浓度为40~60%(w/v)。
[0027]
进一步地,所述的乙醇胺硫酸酯、硫化碱、无机碱和碳酸双(三氯甲)酯的质量比为28:15~25:5~17.5:20;所述的溶剂与乙醇胺硫酸酯的体积质量比为28:60~80(v/w);所述的含碳酸双(三氯甲)酯的溶液中碳酸双(三氯甲)酯浓度为40~50%(w/v);所述的含碳酸双(三氯甲)酯的溶液为碳酸双(三氯甲)酯的水溶液、碳酸双(三氯甲)酯的二氯甲烷溶液、碳酸双(三氯甲)酯的甲苯溶液或碳酸双(三氯甲)酯的异丙醚溶液。
[0028]
进一步地,所述的乙醇胺硫酸酯、硫化碱、无机碱和碳酸双(三氯甲)酯的质量比为28:25:15:20;所述的溶剂与乙醇胺硫酸酯的体积质量比为28:70(v/w);所述的含碳酸双(三氯甲)酯的溶液中碳酸双(三氯甲)酯浓度为40%(w/v)。
[0029]
进一步地,所述的反应温度为0~80℃;优选地,所述的反应温度为20~80℃;更优选地,所述的反应温度为40℃。
[0030]
进一步地,所述的硫化碱为硫化钠、硫化钾、硫氢化钠、硫氢化钾或硫化铵;
[0031]
所述的无机碱为氢氧化钠、氢氧化钾、氢氧化钙、氧化钙、或碱金属或碱土金属的弱酸盐;其中,弱酸盐为碳酸盐、醋酸盐;
[0032]
所述的溶剂为水、二氯甲烷水溶液或甲苯水溶液;其中,二氯甲烷水溶液中水和二氯甲烷的体积比为3~8:1,甲苯水溶液中水和甲苯的体积比为5~10:1。
[0033]
进一步地,所述的硫化碱为硫化钠或硫氢化钠;
[0034]
所述的无机碱为氢氧化钠、氢氧化钾、氢氧化钙或氧化钙;
[0035]
所述的溶剂中,二氯甲烷水溶液中水和二氯甲烷的体积比为6:1;所述甲苯水溶液中水和甲苯的体积比为6:1。
[0036]
进一步地,所述合成方法还包括对反应后的反应液进行提纯,所述提纯包括将反应液过滤,将滤液浓缩得粗品;
[0037]
或,所述提纯包括将反应液过滤,滤液用有机溶剂提取,蒸干有机溶剂,得粗品,所得粗品用80%乙醇水溶液精制,即得。
[0038]
进一步地,所述有机溶剂为二氯甲烷、甲苯。
[0039]
本发明中,w/v代表质量体积比,单位为g/ml。
[0040]
本发明中,v/w代表体积质量比,单位为ml/g。
[0041]
本发明中,“一步法”是指将所有反应原料放在同一反应器内反应,不需将中间体分离出来。使用的是化学反应中常用的“一锅煮”的操作。
[0042]
本发明中,乙醇胺硫酸酯除使用市售试剂外,还可以由硫酸和乙醇胺加热脱水后而得。
[0043]
本发明中,碳酸双(三氯甲)酯为双碳酸酯,csa号是32315-10-9。
[0044]
本发明内容十分简洁,将合成化合物的几种原料一并投入反应器中,原料为乙醇胺、硫酸酯(固体),硫化碱(如硫化钠固体)无机碱(如钠碱、钙碱固体);再加入水和/或有机溶剂,然后将碳酸双(三甲基)酯溶解在有机溶剂中在一定温度下(0-80℃)滴入其中,进行反应,同高效液相测定产品转化率达到要求(如60%-90%)即可停止反应,进一步的将反应液进行过滤,滤去固体反应副产物,滤液可直接蒸发得粗品或用有机溶剂提出后,蒸去有机溶剂后得到同样的粗品。
[0045]
本发明提供了一种氧化氮硫环戊烷化合物2-氧化四氢噻唑1,3的合成方法。该合成方法利用“一锅煮”的方式(一步法)制备2-氧化四氢噻唑1,3,首先利用现有技术中最廉价的办法制备半胱胺,但并不分离出半胱胺,而是在制备半胱胺的同时加入碳酸双(三氯甲)酯,使反应产生半胱胺来不及进行副反应,就立即形成性质稳定的化合物2-氧化四氢噻唑1,3,这样避免了现有技术制备半胱胺转化率低的问题,提高了反应的转化率。同时,本发明化合物2-氧化四氢噻唑1,3的合成方法反应条件十分温和,且主要以水作为溶剂,操作十分简便流畅,对环境友好,产品收率高,纯度高,质量好,价格低廉(所用原料比用半胱胺低一半左右),具有很大的实用价值。
[0046]
显然,根据本发明的上述内容,按照本领域的普通技术知识和惯用手段,在不脱离本发明上述基本技术思想前提下,还可以做出其它多种形式的修改、替换或变更。
[0047]
以下通过实施例形式的具体实施方式,对本发明的上述内容再作进一步的详细说明。但不应将此理解为本发明上述主题的范围仅限于以下的实例。凡基于本发明上述内容所实现的技术均属于本发明的范围。
具体实施方式
[0048]
本发明具体实施方式中使用的原料、设备均为已知产品,通过购买市售产品获得。
[0049]
实施例1、本发明2-氧化四氢噻唑1,3的合成方法
[0050]
取乙醇胺硫酸酯280g、硫化钠250g、氧化钙150g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及二氯甲烷100ml,然后在40℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v:v)精制得产品,收得产品140g,熔点49℃,gc测定含量为98%以上,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0051]
实施例2、本发明2-氧化四氢噻唑1,3的合成方法
[0052]
取硫酸196g、乙醇胺122g,加入2000ml反应瓶中,加热至120℃脱水至干,再加入水800ml、硫化钠250g及氢氧化钠80g,然后在40℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v:v)精制得产品,收得产品142g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0053]
实施例3、本发明2-氧化四氢噻唑1,3的合成方法
[0054]
取乙醇胺硫酸酯280g、硫化钠250g、氢氧化钙150g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及二氯甲烷100ml,然后在40℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸
干后得粗品,用乙醇:水(80:100,v:v)精制得产品,收得产品138g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0055]
实施例4、本发明2-氧化四氢噻唑1,3的合成方法
[0056]
取乙醇胺硫酸酯280g、硫化钠250g、氢氧化钾150g,加入2000ml反应瓶中,并在反应瓶中加入水800ml及二氯甲烷100ml,在40℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v:v)精制得产品,收得产品132g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0057]
实施例5、本发明2-氧化四氢噻唑1,3的合成方法
[0058]
取乙醇胺硫酸酯280g、氢氧化钠50g,加入2000ml反应瓶中,并在反应瓶中加入水800ml,搅拌使其完全溶解,然后加入硫化钠200g,滴加400ml含200g碳酸双(三氯甲)酯的甲苯溶液,再加入氧化钙125g,在20℃反应,用hplc测定反应完毕后,分出有机层,有机层冷冻后析出产品102g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0059]
实施例6、本发明2-氧化四氢噻唑1,3的合成方法
[0060]
取乙醇胺硫酸酯280g、氢氧化钠50g,加入2000ml反应瓶中,并在反应瓶中加入水600ml,搅拌使其完全溶解,然后加入硫化钠200g,滴加400ml含200g碳酸双(三氯甲)酯的甲苯溶液,在80℃反应,用hplc测定反应完毕后,分出有机层,有机层冷冻后析出产品102g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0061]
实施例7、本发明2-氧化四氢噻唑1,3的合成方法
[0062]
取乙醇胺硫酸酯280g、氢氧化钾60g,加入2000ml反应瓶中,并在反应瓶中加入水800ml,搅拌使其完全溶解,然后加入硫化钠200g,滴加400ml含200g碳酸双(三氯甲)酯的异丙醚溶液,在20℃反应,用hplc测定反应完毕后,分出有机层,有机层冷冻后析出产品115g,熔点39.5℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0063]
实施例8、本发明2-氧化四氢噻唑1,3的合成方法
[0064]
取乙醇胺硫酸酯280g、硫化钠250g、氧化钙150g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及二氯甲烷100ml,在40℃滴加500ml含200g碳酸双(三氯甲)酯的水溶液,冷冻得二氯甲烷的溶液。滴加完成后回收有机溶剂,水溶液浓缩至出水800ml后冷至室温过滤,水溶液冷却后用乙醇:水(80:100,v/v)精制得产品,收得产品156g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0065]
实施例9、本发明2-氧化四氢噻唑1,3的合成方法
[0066]
取乙醇胺硫酸酯280g、硫化钠250g、氧化钙150g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及甲苯100ml,在80℃滴加500ml含200g碳酸双(三氯甲)酯的甲苯溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v/v)精制得产品,收得产品140g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0067]
实施例10、本发明2-氧化四氢噻唑1,3的合成方法
[0068]
取乙醇胺硫酸酯280g、硫化钠250g、氢氧化钠80g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及甲苯100ml,在40℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶
液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v/v)精制得产品,收得产品140g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0069]
实施例11、本发明2-氧化四氢噻唑1,3的合成方法
[0070]
取乙醇胺硫酸酯280g、硫氢钠150g、氢氧化钠120g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及二氯甲烷100ml,在30℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v/v)精制得产品,收得产品125g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0071]
实施例12、本发明2-氧化四氢噻唑1,3的合成方法
[0072]
取乙醇胺硫酸酯280g、硫化钠250g、氧化钙150g,加入2000ml反应瓶中,并在反应瓶中加入水600ml及二氯甲烷200ml,在50℃滴加500ml含200g碳酸双(三氯甲)酯的二氯甲烷溶液。滴加完成后回收有机溶剂,反应液过滤,滤液用二氯甲烷提取,将有机层蒸干后得粗品,用乙醇:水(80:100,v/v)精制得产品,收得产品162g,熔点49℃,gc测定含量为大于96%,产品与2-氧化四氢噻唑1,3标准品hplc图谱一致。
[0073]
综上,本发明提供了一种氧化氮硫环戊烷化合物2-氧化四氢噻唑1,3的合成方法。该合成方法利用“一锅煮”的方式(一步法)制备2-氧化四氢噻唑1,3,首先利用现有技术中最廉价的办法制备半胱胺,但并不分离出半胱胺,而是在制备半胱胺的同时加入碳酸双(三氯甲)酯,使反应产生半胱胺来不及进行副反应,就立即形成性质稳定的化合物2-氧化四氢噻唑1,3,这样避免了现有技术制备半胱胺转化率低的问题,提高了反应的转化率。同时,本发明化合物2-氧化四氢噻唑1,3的合成方法反应条件十分温和,且主要以水作为溶剂,操作十分简便流畅,对环境友好,产品收率高,纯度高,质量好,价格低廉(所用原料比用半胱胺低一半左右),具有很大的实用价值。
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1