帕瑞昔布钠遗传毒性杂质及其制备方法与流程
本发明涉及药物合成
技术领域:
,特别是涉及帕瑞昔布钠遗传毒性杂质及其制备方法。
背景技术:
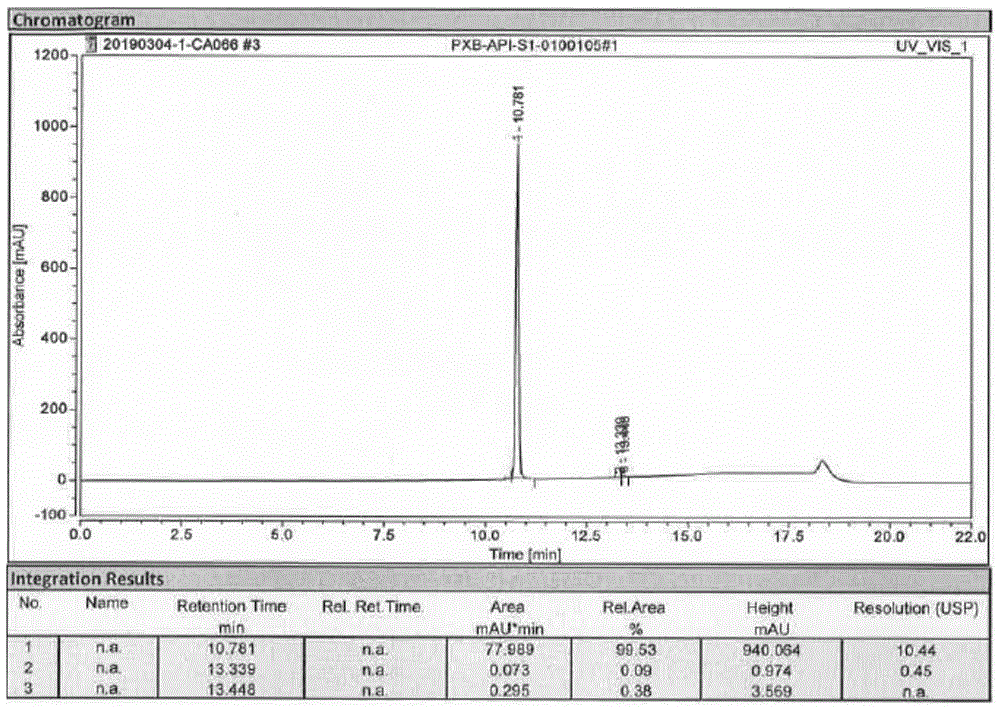
:帕瑞昔布钠是pharmacia公司开发的、全球第一个可静脉和肌肉注射的特异性环氧合酶-2(cox-2)抑制剂,化学名为n-[[4-(5-甲基-3-苯基-4-异恶唑基)苯基]磺酰基]丙酰胺钠盐,结构式如下,商品名“特耐”,2002年初在欧洲上市,2008年进入中国。帕瑞昔布钠是伐地昔布的水溶性前药,注射后在肝脏迅速水解成伐地昔布,临床上主要用于手术后疼痛的短期治疗,也可用于中度或重度术后急性疼痛的治疗,其快速持久镇痛的特性与良好安全性的结合,为术后镇痛提供了更优选择。帕瑞昔布钠的合成路线较多,总结后主要有以下几种:合成路线1:cn97193747.8首次报道了帕瑞昔布钠的合成。该路线以4-(5-甲基-3-苯基-4-异恶唑基)苯磺酰胺(伐地昔布)为起始原料,与丙酸酐发生丙酰化反应后得到帕瑞昔布,再与氢氧化钠成盐得到帕瑞昔布钠。反应方程式如下:合成路线2:wo2005123701采用1,2-二苯乙酮为起始原料,与四氢吡咯缩合,再氯乙酰化;随后在醋酸纳和盐酸羟胺作用下脱水、环合,并在三氟乙酸催化下脱水得到5-甲基-3,4-二苯基异恶唑;再进行氯磺化、氨化反应生成伐地昔布;又经过丙酰化得到帕瑞昔布,最后与氢氧化钠成盐得到终产品帕瑞昔布钠。反应方程式如下:合成路线3:ep1550658则以1-苯基丙酮为起始原料,与四氢吡咯缩合,再与苯腈n氧化物反应成环,在浓盐酸中脱四氢吡咯,形成双键得到5-甲基-3,4-二苯基异恶唑;再进行氯磺化、氨化反应生成伐地昔布;又经过丙酰化得到帕瑞昔布,最后与氢氧化钠成盐得到终产品帕瑞昔布钠。反应方程式如下:合成路线4:wo2003029230采用1,2-二苯乙酮为起始原料,与羟胺反应生成二苯乙酮肟,在正己基锂/乙酸乙酯作用下关环;又在三氟乙酸催化下脱水形成5-甲基-3,4-二苯基异恶唑,再进行氯磺化、氨化反应生成伐地昔布;又经过丙酰化得到帕瑞昔布,最后与氢氧化钠成盐得到终产品帕瑞昔布钠。反应方程式如下:分析上述合成路线可知,伐地昔布是帕瑞昔布钠的关键中间体,而伐地昔布一般是由5-甲基-3,4-二苯基异恶唑先与氯磺酸发生氯磺化反应,生成中间体氯磺化物,再与浓氨水发生氨化反应得到伐地昔布。目前在从伐地昔布经丙酰化和成盐这两步反应制备终产品帕瑞昔布钠的合成路线中,每步反应或随后的精制纯化操作均多次用到醇类溶剂,如:甲醇、乙醇、异丙醇等。氨化反应中未消耗完全的氯磺化物,会残留在氨化反应步骤的产物伐地昔布中,与后续使用的醇类溶剂,反应生成如下3个芳基磺酸酯杂质:4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯。磺酸酯为业内公认的警示结构,根据《人用药品注册技术要求国际协调会议(ich)》的文件m7,即《评估和控制药物中dna反应性(致突变)杂质以限制潜在的致癌风险》中的杂质分类规则,和《遗传毒性杂质控制指导原则-中国药典2020版征求意见稿》的规定,上述3个芳基磺酸酯杂质系3类遗传毒性杂质,须参照毒理学关注阈值(ttc)控制其允许每日摄入量(adi)在1.5μg/天。因帕瑞昔布钠的最大日剂量为80mg/天,故这3个芳基磺酸酯杂质的限度计算如下:限度(%)=1.5μg/80mg=0.0018%=18ppm。由此可见,在帕瑞昔布钠原料药的日常出厂放行工作中,上述3个控制限度极低的遗传毒性杂质的定性检测和定量检测,对分析部门构成了巨大挑战,因此首先就需要开发出便捷的高纯度对照品的制备方法。对照品纯度越高,体现在液/气相色谱或薄层色谱上的主峰或主斑点就越明确,限度极低的遗传毒性杂质的定性检测和定量检测的结果也就越准确;对照品的制备操作程序越便捷,收率越高,则越容易快速满足分析部门的要求。只有能及时供应合乎纯度要求的对照品,才能确保后续分析部门顺利开展液相或气相检测方法的研究工作。专利201510843521.8公开了一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯的制备方法:从5-甲基-3,4-二苯基异恶唑出发,先发生氯磺化反应得到氯磺化物中间体(对位),然后与乙醇/吡啶发生酯化反应,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯。反应方程式如下:然而,该方法在第一步氯磺化反应中,副反应较多,增加了第二步酯化反应产物的分离纯化难度,而且,在第二步酯化反应中,为了顺利完成酯化反应,加入缚酸剂吡啶,增加了引入新有机杂质的风险,也增加了清除的难度,此外,该方法将得到的粗品潮品精制的过程中,极易引起产物4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯的水解。总的来说,专利201510843521.8制得的4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯,无论是从产品的纯度或收率上来说,都不甚理想。技术实现要素:基于此,本发明提供一种帕瑞昔布钠遗传毒性杂质的制备方法,制备了3种帕瑞昔布钠遗传毒性杂质,分别为4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯,制得的上述3种帕瑞昔布钠遗传毒性杂质纯度可以达到99.5%以上,甚至是99.73%;收率可以达到67%以上,甚至是71.0%。具体技术方案为:一种帕瑞昔布钠遗传毒性杂质的制备方法,包括以下步骤:于温度-5℃~0℃下,混合5-甲基-3,4-二苯基异恶唑和二氯甲烷,再以<3ml/分钟的速率加入氯磺酸,所述氯磺酸加入完毕后,升温至35±5℃,采用高效液相色谱法监控反应进程,按面积归一法计算,至反应体系中所述5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,得反应液;向所述反应液中加入冰水淬灭,萃取,得含有对位氯磺化物中间体的二氯甲烷溶液;向所述含有对位氯磺化物中间体的二氯甲烷溶液中加入甲醇钠的甲醇溶液、乙醇钠的乙醇溶液或异丙醇钠的异丙醇溶液,反应2h-4h,除去不溶性物质和二氯甲烷,于0℃±5℃结晶,得4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯或4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯。本发明还提供一种帕瑞昔布钠遗传毒性杂质。具体技术方案为:一种帕瑞昔布钠遗传毒性杂质,由上述制备方法制得。与现有技术相比,本发明具有以下有益效果:本发明所述的制备方法在5-甲基-3,4-二苯基异恶唑与氯磺酸发生氯磺化反应的过程中,通过控制氯磺酸的加入速率,减少邻位、间位异构体,甚至减少在另一个苯环上发生取代反应形成新的异构体的发生。同时采用高效液相色谱法监控反应体系中5-甲基-3,4-二苯基异恶唑和间位氯磺化物异构体的含量,进一步限制能够进入到酯化反应的副产物的含量,减少酯化产物分离纯化的难度。此外,在酯化反应中,通过向含有对位氯磺化物中间体的二氯甲烷溶液中加入甲醇钠的甲醇溶液、乙醇钠的乙醇溶液或异丙醇钠的异丙醇溶液,完成酯化,无需使用缚酸剂,避免了引入新的有机物的风险,同时,本发明所述酯化反应常温下即可进行,还能避免酯化产物高温下分解。在上述多种手段的共同作用下,使本发明所述制备方法制得的3种帕瑞昔布钠遗传毒性杂质纯度可以达到99.5%以上,甚至是99.73%;收率可以达到67%以上,甚至是71.0%。可作为帕瑞昔布钠原料质量控制的杂质对照品。附图说明图1为实施例1中4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯的高效液相色谱图;图2为实施例2中4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯的高效液相色谱图;图3为实施例3中4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯的高效液相色谱图;图4为对比例1中4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯的高效液相色谱图;图5为对比例2中4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯的高效液相色谱图。具体实施方式以下结合具体实施例对本发明进一步详细的说明。本发明可以以许多不同的形式来实现,并不限于本文所描述的实施方式。相反地,提供这些实施方式的目的是使对本发明公开内容理解更加透彻全面。除非另有定义,本文所使用的所有的技术和科学术语与属于本发明的
技术领域:
的技术人员通常理解的含义相同。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。本文所使用的术语“和/或”包括一个或多个相关的所列项目的任意的和所有的组合。一种帕瑞昔布钠遗传毒性杂质的制备方法,包括以下步骤:于温度-5℃~0℃下,混合5-甲基-3,4-二苯基异恶唑和二氯甲烷,再以<3ml/分钟的速率加入氯磺酸,所述氯磺酸加入完毕后,升温至35±5℃,采用高效液相色谱法监控反应进程,按面积归一法计算,至反应体系中所述5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,得反应液;向所述反应液中加入冰水淬灭,萃取,得含有对位氯磺化物中间体的二氯甲烷溶液;向所述含有对位氯磺化物中间体的二氯甲烷溶液中加入甲醇钠的甲醇溶液、乙醇钠的乙醇溶液或异丙醇钠的异丙醇溶液,反应2h-4h,除去不溶性物质和二氯甲烷,于0℃±5℃结晶,得4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯、4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯或4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯。上述制备方法的反应方程式如下:优选地,氯磺化反应中,所述5-甲基-3,4-二苯基异恶唑和氯磺酸的重量比为1:(3~4)。如果氯磺酸投料量超过这个范围,会引起后处理淬灭的工作量加大和危险性增加,也不利于环保。可以理解地,混合5-甲基-3,4-二苯基异恶唑和二氯甲烷后,连接尾气吸附装置,再加入氯磺酸。在5-甲基-3,4-二苯基异恶唑与氯磺酸发生氯磺化反应的过程中,通过控制氯磺酸的加入速率,减少邻位、间位异构体,甚至减少在另一个苯环上发生取代反应形成新的异构体的发生。所述氯磺酸的加入速率<3ml/分钟。缓慢滴加氯磺酸,控制温度最高不超过25℃。通过控温,也可以避免剧烈反应,减少异构体的产生。可以理解地,氯磺酸滴加完毕后,可以先搅拌0.5小时,然后再升温至35℃±5℃。同时,在氯磺化的反应过程中,采用高效液相色谱法(hplc)监控反应体系中5-甲基-3,4-二苯基异恶唑和间位氯磺化物异构体的含量,进一步限制能够进入到酯化反应的副产物的含量,减少酯化产物分离纯化的难度。优选地,采用高效液相色谱法监控反应进程,按面积归一法计算,至反应体系中所述5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,得反应液。此过程一般耗时8h左右。可以理解地,将所述反应液冷却的方法可以是将反应液缓慢滴入冰水中,控制温度最高不超过15℃,滴加完后搅拌0.5小时。缓慢滴入和控温的目的是避免氯磺酸与水剧烈反应。冷却后,以二氯甲烷为萃取剂,进行萃取,合并有机相,可以用氯化钠水溶液洗涤,还可以用无水硫酸钠干燥,抽滤后,得到含有对位氯磺化物中间体的二氯甲烷溶液。向所述含有对位氯磺化物中间体的二氯甲烷溶液中加入甲醇钠的甲醇溶液、乙醇钠的乙醇溶液或异丙醇钠的异丙醇溶液进行酯化反应,生成相应的磺酸酯和氯化钠。酯化反应过程中,无需使用缚酸剂,避免了引入新的有机物的风险,同时,本发明所述酯化反应常温下即可进行,还能避免酯化产物高温下分解。优选地,所述甲醇钠的甲醇溶液中,甲醇钠与所述5-甲基-3,4-二苯基异恶唑的摩尔比为(1.0~1.1):1,甲醇与所述5-甲基-3,4-二苯基异恶唑的重量比为(2~3):1。优选地,所述乙醇钠的乙醇溶液中,乙醇钠与所述5-甲基-3,4-二苯基异恶唑的摩尔比为(1.0~1.1):1,乙醇与所述5-甲基-3,4-二苯基异恶唑的重量比为(2~3):1。优选地,所述异丙醇钠的异丙醇溶液中,异丙醇钠与所述5-甲基-3,4-二苯基异恶唑的摩尔比为(1.0~1.1):1,异丙醇与所述5-甲基-3,4-二苯基异恶唑的重量比为(2~3):1。可以理解地,上述酯化反应中,可采用薄层色谱法监控反应进程,至反应体系中所述对位氯磺化物中间体消耗完全,此过程一般耗时2h-4h,可以通过抽滤的方法除去不溶性物质,还可通过减压除去二氯甲烷。优选地,采用薄层色谱法监控反应进程时,展开剂为体积比为1:4的二氯甲烷和石油醚的混合溶液。酯化反应后,于0℃~5℃结晶。优选地,结晶的时间为3h-4h。可以理解地,结晶后抽滤,用适量的相对应的醇溶剂对产品进行洗涤,控制温度≤45℃,减压干燥6小时,得到相应的磺酸酯目标物。在上述多种手段的共同作用下,使本发明所述制备方法制得的3种帕瑞昔布钠遗传毒性杂质纯度可以达到99.5%以上,甚至是99.73%;收率可以达到67%以上,甚至是71.0%。可作为帕瑞昔布钠原料质量控制的杂质对照品。上述方法制得的3种帕瑞昔布钠遗传毒性杂质结构式如下:以下结合具体实施例做进一步说明。实施例1本实施例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯及其制备方法,步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以2ml/分钟的速率,缓慢滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用hplc监控反应进程,按面积归一法计算,至起始原料5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入预先配好的乙醇钠的乙醇溶液,其中,乙醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1,乙醇钠与起始原料5-甲基-3,4-二苯基异恶唑的摩尔比为1.05:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,至氯磺化物几乎消耗完全,先抽滤除去不溶性物质,然后室温下稍稍减压除去溶剂二氯甲烷。随后将得到的溶液冷却至0℃,搅拌结晶3小时,抽滤,将固体用适量无水乙醇洗涤后,控制温度≤45℃,减压干燥6小时,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯,收率71.0%。对上述4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯进行hplc测定,得色谱图如图1所示,色谱纯度为99.53%(面积归一化法)。具体测定方法为:色谱柱:sunfirec18,5um,4.6x150mm流动相:a:acnb:0.05%tfainh2o流速:2.5ml/min波长:254nm洗脱梯度:时间(min)a%b%0.0070302.00703012.00208015.00208018.00703023.007030结构确证:高分辨质谱:344.0949(m+h)+。核磁解析如下:核磁氢谱解析数据表核磁碳谱解析数据表实施例2本实施例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯及其制备方法,步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以2ml/分钟的速率,缓慢滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用hplc监控反应进程,按面积归一法计算,至起始原料5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入预先配好的甲醇钠的甲醇溶液,其中,甲醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1,甲醇钠与起始原料5-甲基-3,4-二苯基异恶唑的摩尔比为1.05:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,至氯磺化物几乎消耗完全,先抽滤除去不溶性物质,然后室温下稍稍减压除去溶剂二氯甲烷。随后将得到的溶液冷却至0℃,搅拌结晶3小时,抽滤,将固体用适量无水甲醇洗涤后,控制温度≤45℃,减压干燥6小时,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯,收率68.4%。对上述4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸甲酯进行hplc测定,得色谱图如图2所示,色谱纯度为99.73%(面积归一化法)。测定方法同实施例1。结构确证:高分辨质谱:330.0792(m+h)+。核磁解析如下:核磁氢谱解析数据表核磁碳谱解析数据表实施例3本实施例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯及其制备方法,步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以2ml/分钟的速率,缓慢滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用hplc监控反应进程,按面积归一法计算,至起始原料5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入预先配好的异丙醇钠的异丙醇溶液,其中,异丙醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1,异丙醇钠与起始原料5-甲基-3,4-二苯基异恶唑的摩尔比为1.05:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,至氯磺化物几乎消耗完全,先抽滤除去不溶性物质,然后室温下稍稍减压除去溶剂二氯甲烷。随后将得到的溶液冷却至0℃,搅拌结晶3小时,抽滤,将固体用适量无水异丙醇洗涤后,控制温度≤45℃,减压干燥6小时,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯,收率67.5%。对上述4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸异丙酯进行hplc测定,得色谱图如图3所示,色谱纯度为99.56%(面积归一化法)。测定方法同实施例1。结构确证:高分辨质谱:358.1105(m+h)+。核磁解析如下:核磁氢谱解析数据表核磁碳谱解析数据表对比例1本对比例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯及其制备方法,与实施例1的步骤基本相同,区别仅在于,氯磺酸的加入速率不同,具体步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以5ml/分钟的速率滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用hplc监控反应进程,按面积归一法计算,至起始原料5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入预先配好的乙醇钠的乙醇溶液,其中,乙醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1,乙醇钠与起始原料5-甲基-3,4-二苯基异恶唑的摩尔比为1.05:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,至氯磺化物几乎消耗完全,先抽滤除去不溶性物质,然后室温下稍稍减压除去溶剂二氯甲烷。随后将得到的溶液冷却至0℃,搅拌结晶3小时,抽滤,将固体用适量无水乙醇洗涤后,控制温度≤45℃,减压干燥6小时,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯,收率68.2%。对上述4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯进行hplc测定,得色谱图如图4所示,色谱纯度为93.28%(面积归一化法)。测定方法同实施例1。对比例2本对比例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯及其制备方法,与实施例1的步骤基本相同,区别仅在于,磺化反应时,采用薄层色谱法代替高效液相色谱法,监控反应的进行。具体步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以2ml/分钟的速率,缓慢滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用薄层色谱法监控反应进程(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm),至起始原料5-甲基-3,4-二苯基异恶唑几乎消耗完全,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入预先配好的乙醇钠的乙醇溶液,其中,乙醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1,乙醇钠与起始原料5-甲基-3,4-二苯基异恶唑的摩尔比为1.05:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,至氯磺化物几乎消耗完全,先抽滤除去不溶性物质,然后室温下稍稍减压除去溶剂二氯甲烷。随后将得到的溶液冷却至0℃,搅拌结晶3小时,抽滤,将固体用适量无水乙醇洗涤后,控制温度≤45℃,减压干燥6小时,得到4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯,收率68.7%。对上述4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯进行hplc测定,得色谱图如图5所示,色谱纯度为93.29%(面积归一化法)。测定方法同实施例1。对比例3本对比例提供一种4-(5-甲基-3-苯基-4-异恶唑基)苯磺酸乙酯及其制备方法,与实施例1的步骤基本相同,区别仅在于,酯化反应中,只加入乙醇,未加入乙醇钠。具体步骤如下:向100ml三口反应瓶中加入50g二氯甲烷和5g起始原料5-甲基-3,4-二苯基异恶唑,搅拌降温至-2℃,连接尾气吸附装置,以2ml/分钟的速率,缓慢滴加16.5g氯磺酸,控制反应温度最高不超过25℃。滴加完后先搅拌0.5小时,然后升温至35℃,采用hplc监控反应进程,按面积归一法计算,至起始原料5-甲基-3,4-二苯基异恶唑≤1.0%、间位氯磺化物异构体≤1.0%,停止反应,得反应液。将反应液缓慢滴入到冰水中,冷却,控制温度最高不超过15℃,滴加完后搅拌0.5小时。静置分液,用新鲜二氯甲烷萃取,然后合并有机相,再用氯化钠水溶液洗涤并分液,无水硫酸钠干燥。抽滤,得到对位氯磺化物中间体的二氯甲烷溶液,待用。室温下,向上述对位氯磺化物中间体的二氯甲烷溶液中,加入溶剂乙醇,其中,乙醇与起始原料5-甲基-3,4-二苯基异恶唑的重量比为2:1。搅拌反应3小时后,采用tlc(展开剂为二氯甲烷:石油醚=1:4,v:v,254nm)监控反应进程,结果显示,两者未发生反应。以上所述实施例的各技术特征可以进行任意的组合,为使描述简洁,未对上述实施例中的各个技术特征所有可能的组合都进行描述,然而,只要这些技术特征的组合不存在矛盾,都应当认为是本说明书记载的范围。以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对本发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。因此,本发明专利的保护范围应以所附权利要求为准。当前第1页1 2 3
相关技术
网友询问留言
已有0条留言
- 还没有人留言评论。精彩留言会获得点赞!
1