一种苯并噻唑衍生物及其制备方法和白酒酒精度快速可视化识别的应用与流程
本发明涉及一种苯并噻唑衍生物及其制备方法和白酒酒精度快速可视化识别的应用,属于荧光材料技术领域。
背景技术:
文献已报道的检测水分含量的方法或材料主要包括以下几种:
1)卡尔·费休滴定法:利用碘氧化二氧化硫时,需要一定量的水参加反应,从而可以用于测定水分含量的绝对值,适用于大部分固态和液态样品,具有检测范围广,准确度高等优点,但是配置的费休试剂需要绝对隔离空气和水分,很不稳定,难以保存;
2)高分辨率电容型水分传感器:可检测多种有机溶剂中的痕量水分,在乙醇中检测限为0.019%(w/w),响应时间为60ms,但传感器的灵敏度受温度的干扰大;
3)利用有机聚合物、金属配合物、金属有机框架(mof)、无机钙钛矿等材料制备的检测水分含量的荧光传感材料,例如:利用仿生矿化的方法合成的poly-lnmofs探针,不同水分含量(包括痕量)能够引起荧光强度发生线性关系变化,从而根据stern-volmer方程i0/i=1+ksv[h2o]计算未知的有机溶液中的水分含量,该方法不能进行实时原位检测。
由此可见,传统的检测水分含量的方法都存在明显的缺陷,难以大面积推广应用。
近年来,也有使用合成的有机小分子作为检测水分含量的荧光传感器,例如:基于罗丹明的有机溶剂痕量水分传感器,可以在液态和试纸条上检测水分含量,检测范围为1%~8%,在dmf中检测限为0.026%;基于吡喃酮和三苯胺的有机溶剂痕量水分传感器,在不同水分含量中荧光强度线性减弱,通过stern-volmer方程计算水分含量,其中在四氢呋喃中检测范围为0~10%,检测限为0.0096%。然而,上述的水分含量检测方法普遍存在对操作要求高、不易携带、样品制备复杂、测定结果不直观、检测范围小等不足。
因此,有必要开发一种兼具成本低廉、合成简单、分析方便、原位检测、无环境干扰、大范围、可视化检测等优点的水分含量检测材料。
技术实现要素:
本发明的目的在于提供一种苯并噻唑衍生物及其制备方法和白酒酒精度快速可视化识别的应用。
本发明所采取的技术方案是:
一种苯并噻唑衍生物,其结构式为:
上述苯并噻唑衍生物的制备方法包括以下步骤:
1)进行5-溴间苯二甲酸和邻氨基苯硫酚的反应,得到
2)进行
优选的,上述苯并噻唑衍生物的制备方法包括以下步骤:
1)将5-溴间苯二甲酸、邻氨基苯硫酚和多聚磷酸加入反应釜,160~180℃反应24~48h,进行产物分离和纯化,得到
2)将
优选的,步骤1)所述5-溴间苯二甲酸、邻氨基苯硫酚的摩尔比为1:(2.0~2.4)。
优选的,步骤1)所述产物分离和纯化的具体操作为:反应结束后将产物冷却至室温,用naoh水溶液调节ph至9~10,有固体析出,减压抽滤,用四氢呋喃对抽滤得到的固体进行重结晶。
优选的,步骤2)所述
优选的,步骤2)所述钯催化剂为pb(pph3)2cl2、pdcl2、pd(ch3co2)2中的至少一种。
优选的,步骤2)所述铜催化剂为cui、cucl、cubr中的至少一种。
优选的,步骤2)所述产物分离和纯化的具体操作为:反应结束后将产物冷却至室温,加入饱和的氯化铵溶液猝灭反应,用二氯甲烷和饱和氯化铵溶液进行萃取,收集有机相进行干燥除水、旋干溶剂,进行柱层析分离。
一种酒精度传感器,包含上述苯并噻唑衍生物。
本发明的有益效果是:本发明的苯并噻唑衍生物属于d-π-a型荧光材料,水溶性和发光性能良好,且其合成工艺简单、制造成本低廉,可以用于白酒酒精度的快速可视化识别。
1)本发明的苯并噻唑衍生物以廉价易得的5-溴间苯二甲酸、邻氨基苯硫酚、4-乙炔基苯甲醚等为原料通过两步合成法制备,不需要定制的合成仪器,反应容易控制,产物提纯简单,产率可达60%;
2)本发明的苯并噻唑衍生物在液态时检测的酒精度(vol)范围为30%~60%,在滤纸条上检测的酒精度范围为40%~50%,可以用于白酒酒精度的快速可视化识别。
附图说明
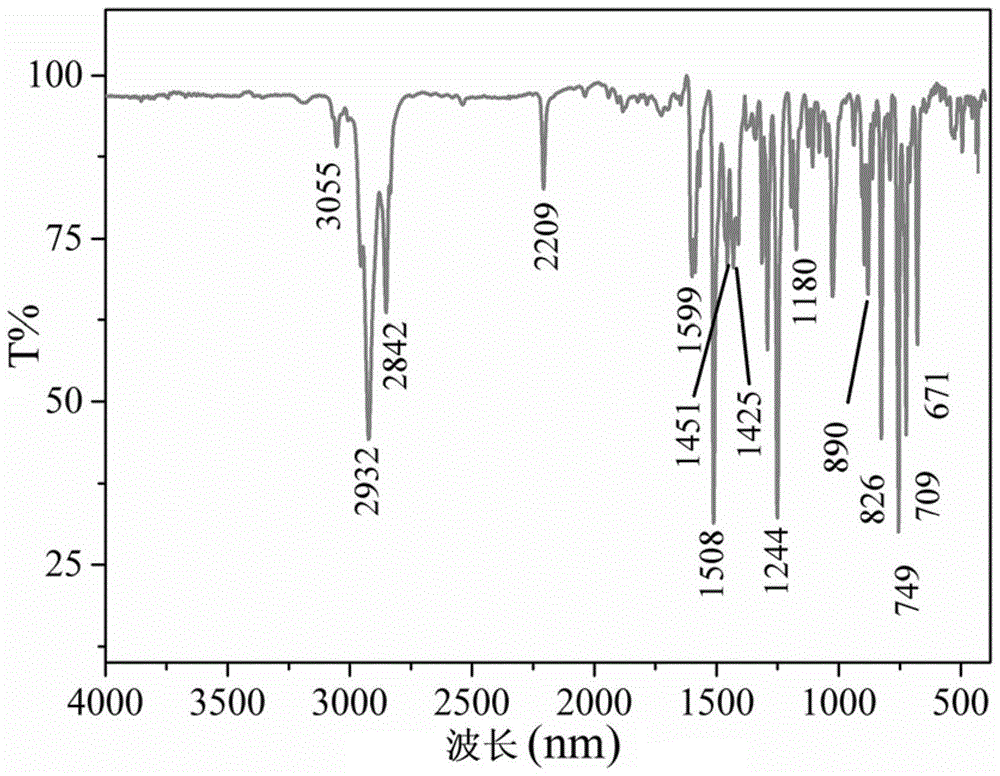
图1为苯并噻唑衍生物的ft-ir图谱。
图2为苯并噻唑衍生物的1hnmr图谱。
图3为苯并噻唑衍生物的13cnmr图谱。
图4为苯并噻唑衍生物的荧光性能测试(液态)的显色照片。
图5为苯并噻唑衍生物的荧光性能测试(试纸)的显色照片。
具体实施方式
下面结合具体实施例对本发明作进一步的解释和说明。
实施例:
一种苯并噻唑衍生物,其制备方法包括以下步骤:
1)将2mmol的5-溴间苯二甲酸、4.4mmol的邻氨基苯硫酚和15ml的多聚磷酸加入50ml的圆底烧瓶中,升温至170℃反应36h,将反应物冷却至室温,用naoh水溶液调节ph至9,有大量的固体析出,减压抽滤,用四氢呋喃对抽滤得到的固体进行重结晶,得到
2)将0.1mmol的
苯并噻唑衍生物的合成路线如下所示:
苯并噻唑衍生物的ft-ir图谱(kbr,ν,cm-1)如图1所示。
由图1可知:
3055cm-1处为芳香环上的不饱和c-h的伸缩振动吸收峰;2932cm-1和2842cm-1处为饱和c-h的伸缩振动吸收峰;2209cm-1处为不饱和c≡c键的伸缩振动吸收峰;1599cm-1、1508cm-1、1451cm-1和1425cm-1处为芳香环骨架伸缩振动吸收峰;1244cm-1处为c-n键的伸缩振动吸收峰;1180cm-1处为c-s键的伸缩振动吸收峰;890cm-1处为苯环孤立氢;709cm-1处为苯环1,3,5-三取代;826cm-1处为苯环1,4-二取代;749cm-1处为苯环1,2-二取代;
综上可知,
苯并噻唑衍生物的1hnmr图谱如图2所示,13cnmr图谱如图3所示。
由图2和图3可知(解谱分析):
1hnmr(cdcl3,600mhz):δ=3.85(s,3h,och3-18),δ=6.92(d,j=6.0hz,2h,arh-2,2′),7.40-7.45(m,2h,arh-13,13′),7.51-7.55(m,4h,arh-3,3′,14,14′),7.94(d,j=6.0hz,2h,arh-12,12′),8.13(d,j=6.0hz,2h,arh-15,15′),8.32(s,2h,arh-8,8′),8.70(s,1h,arh-17);
13cnmr(cdcl3,150mhz):δ=55.3(c-18),86.6(c-5),91.6(c-6),114.1(c-2,2′),114.7(c-4),121.8(c-15,15′),123.5(c-12,12′),125.5(c-7),125.6(c-13,13′,17),126.6(c-14,14),132.1(c-8,8′),133.3(c-3,3′),134.7(c-9,9′),135.2(c-11,11′),154.0(c-16,16′),160.0(c-1),166.2(c-10,10′)。
性能测试:
1)苯并噻唑衍生物的荧光性能测试(液态):
选取11个1.5ml的一次性离心管,分别加入1ml酒精度(vol)为0、10%、20%、30%、40%、50%、60%、70%、80%、90%和100%的酒精溶液,再在离心管中各加入50μl浓度10-3mol/l的苯并噻唑衍生物溶液,盖紧盖子,震荡10s,再将离心管置于波长365nm的紫外灯下,得到的显色照片如图4所示。
由图4可知:酒精度(vol)为30%~60%的混合溶液呈现明显的红橙色荧光,说明本发明的苯并噻唑衍生物能够在液态有效识别常见的酒精度。
2)苯并噻唑衍生物的荧光性能测试(试纸):
裁剪11块大小相同的空白滤纸条,空白滤纸条置于波长365nm的紫外灯下得到的显色照片如图5中的a所示;再选取11个1.5ml的一次性离心管,分别加入1ml酒精度(vol)为0、10%、20%、30%、40%、50%、60%、70%、80%、90%和100%的酒精溶液,再在离心管中各加入50μl浓度10-3mol/l的苯并噻唑衍生物溶液,盖紧盖子,震荡10s,再从离心管中分别取3滴溶液滴加到裁剪好的11块大小相同的空白滤纸条上,等待1min,再将滤纸条置于波长365nm的紫外灯下,得到的显色照片如图5中的b所示。
由图5可知:酒精度(vol)为30%~70%时滤纸条上显示出橙红色荧光,尤其是在酒精度为40%~50%时滤纸条上的橙红色荧光最亮,说明本发明的苯并噻唑衍生物可以应用于快速准确识别酒精度为40%~50%的商业白酒。
上述实施例为本发明较佳的实施方式,但本发明的实施方式并不受上述实施例的限制,其他的任何未背离本发明的精神实质与原理下所作的改变、修饰、替代、组合、简化,均应为等效的置换方式,都包含在本发明的保护范围之内。
- 还没有人留言评论。精彩留言会获得点赞!